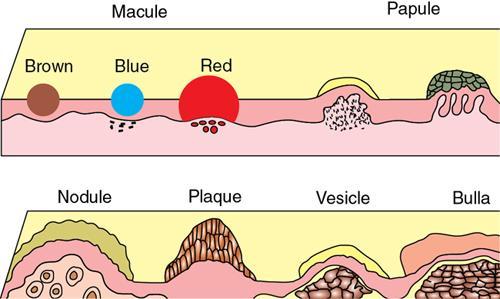
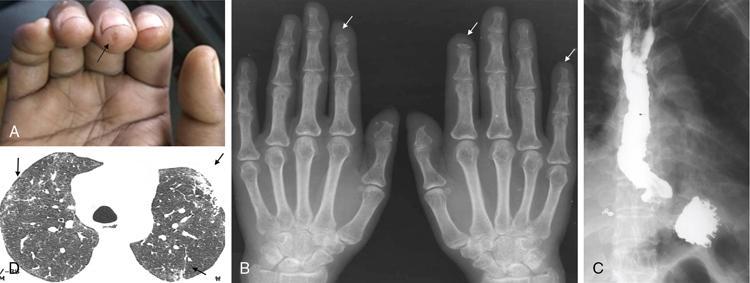
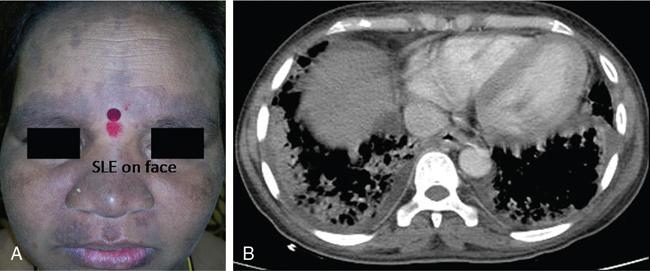
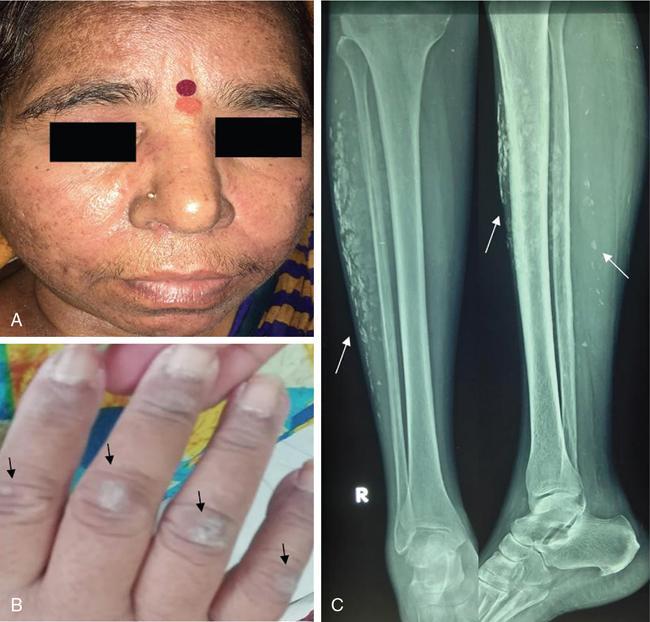
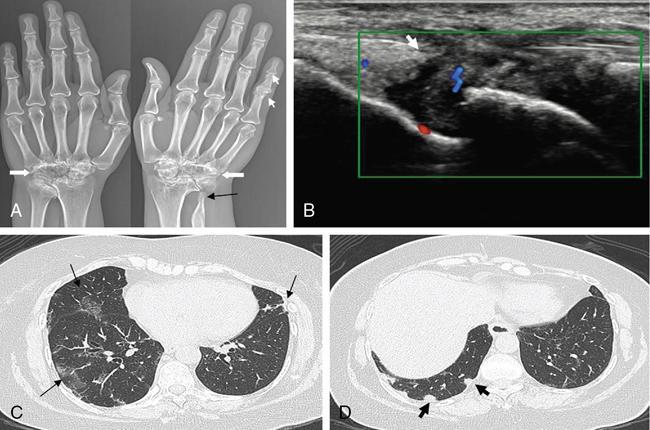
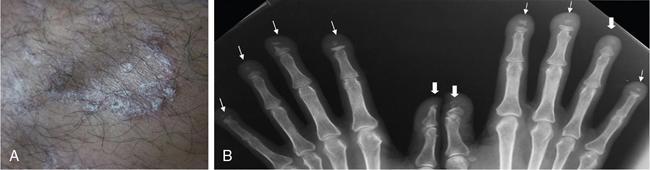
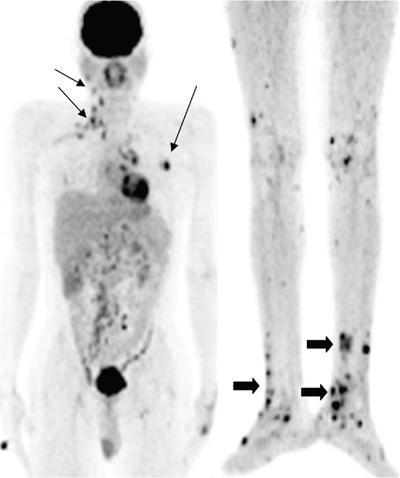
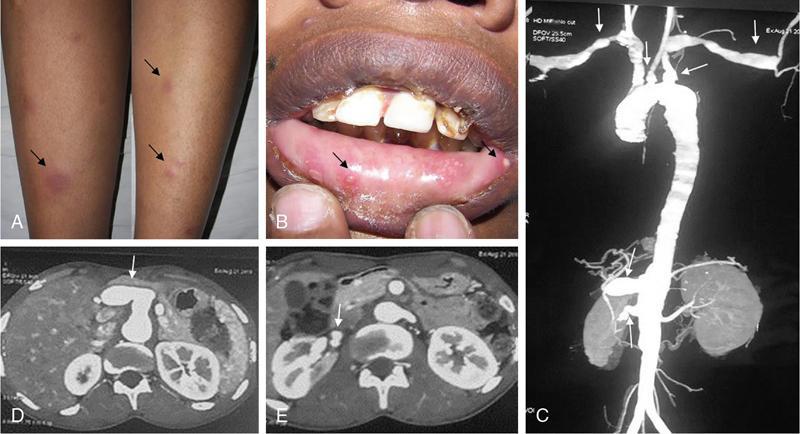
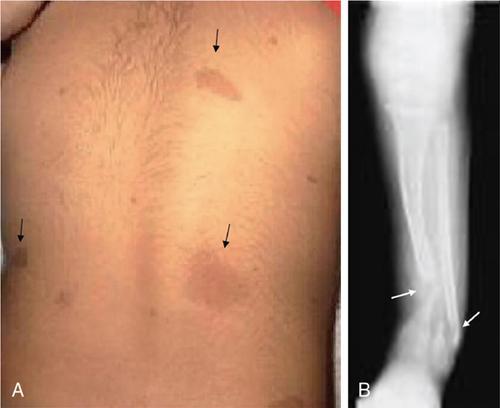
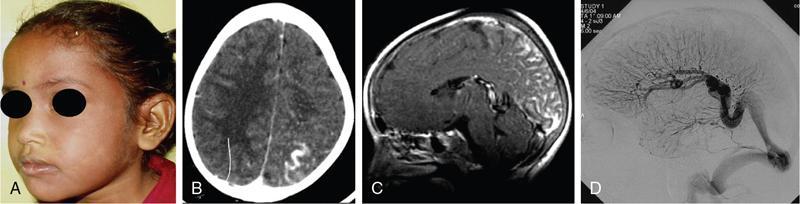
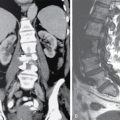
DERMATOLOGICAL MANIFESTATIONS IN SYSTEMIC DISEASES – ROLE OF IMAGING Anitha Mandava, Veeraiah Koppula The skin and its appendages (nail, hair follicles, sebaceous glands, apocrine and eccrine sweat glands) together constitute the largest organ in the human body. In addition to acting as a protective barrier against many harmful external agents, the skin also performs several distinct functions such as maintaining constant core body temperature by thermoregulation, synthesizing vitamin D through a biochemical action, providing insulation and immunological surveillance and perceiving the sense of touch, pain, temperature and deep pressure. Skin also has psychosocial and aesthetic functions and usually reflects the general health of a person. Skin ailments account for one-fourth of human diseases and approximately one-third of the world’s population are being afflicted by at least one ailment or the other during their lifetime. Very often, skin lesions are an initial manifestation of systemic disorder, and in some cases, a characteristic dermatologic feature can either suggest a specific diagnosis or provide clues to establish accurate diagnosis. In some diseases, the cutaneous lesions can herald an internal abnormality including malignancy, and in few disorders, the severity of skin disease may reflect and correlate with the underlying systemic disease. Most of the cutaneous lesions are readily visible and easily accessible for clinical examination and diagnostic procedures without any special instruments. In multisystemic diseases, biopsies can be performed from skin lesions instead of internal organs without causing much distress and morbidity to the patients. Dermatologic manifestations occur in several hereditary syndromes, autoimmune, rheumatologic, hematologic, gastrointestinal, renal, cardiac and pulmonary diseases, infections and internal malignancies. Genodermatoses are inherited disorders occurring due to genetic alterations or mutations with cutaneous and other systemic manifestations. Given the myriad diseases presenting with cutaneous manifestations, a detailed account of all of them is beyond the purview of this book. In this chapter, an overview of important systemic conditions associated with dermatological manifestations is presented. Various types of cutaneous lesions are described in association with systemic diseases, which include erosion, macule, papule, pustule, vesicle, nodule, tumour, bulla, cyst, plaque, patch, ulcer and wheal. Vascular abnormalities are seen as erythema, petechiae, purpura and telangiectasias (Fig. 2.14.1.1, Table 2.14.1.1). Many of the systemic diseases are associated with focal or diffuse skin discoloration, hypopigmentation, hyperpigmentation, photosensitivity, abnormalities of hair and nails such as alopecia, hirsutism, onycholysis and discoloration of nails. The location, history of evolution and progression of lesions, duration (acute or chronic), family history, drug history and associated systemic symptoms such as fever and arthralgias are important during the initial evaluation and diagnosis. Several systemic diseases have cutaneous manifestations due to the involvement of multiple systems by the same pathological process. There is significant overlap in the classification of multisystemic, connective tissue and autoinflammatory disorders with cutaneous manifestations. The underlying pathology affecting multiple systems includes abnormalities and deficiencies of innate immune system, abnormal lymphocyte or antigen–antibody mediated immune response, autoimmune and autoinflammatory diseases, disorders secondary to the activation of chemokine, cytokine and complement pathways and crystallopathies. The common collagen or connective tissue diseases and rheumatic disorders with cutaneous manifestations include progressive systemic sclerosis (PSS), systemic lupus erythematosus (SLE), dermatomyositis (DM), rheumatoid arthritis (RA), relapsing polychondritis, gout, hemochromatosis, seronegative spondyloarthropathies such as ankylosing spondylitis (AS), psoriasis, Reiter’s syndrome, juvenile rheumatoid arthritis (JRA), Behcet’s syndrome and enterospondylarthritis (Crohn’s disease, ulcerative colitis). Progressive systemic sclerosis (PSS) or scleroderma is a rare disease that can affect virtually any organ and is characterized by excessive collagen deposition in the skin and various other organs. Bilateral symmetrical skin thickening, the hallmark of PSS, differentiates it from other connective tissue diseases, and the presence of sclerodactyly, Raynaud’s phenomenon, nail fold capillary changes, fingertip lesions, and telangiectasias distinguishes it from localized scleroderma (morphea). The acronym “CREST” syndrome refers to the presence of calcinosis cutis, Raynaud’s phenomenon, esophageal dysmotility, sclerodactyly and telangiectasia. In 40% of cases, dystrophic calcifications are seen in the skin and subcutaneous soft tissues (calcinosis cutis). The American College of Rheumatology (ACR) criteria for the diagnosis of PSS include the presence of characteristic skin manifestations, cardiopulmonary manifestations and scleroderma specific autoantibodies. The cutaneous manifestations of PSS include bilateral symmetrical skin thickening (sclerosis) of fingers of both hands, advancing from distal to proximal extremities, puffy fingers, sclerodactyly, vascular manifestations, fingertip lesions (digital ulcers and pitting scars) and vitiligo-like hypopigmentation or hyperpigmentation. Systemic manifestations of PSS include the musculoskeletal abnormalities: soft-tissue swelling, puffy fingers, joint contractures, acroosteolysis (resorption of the terminal phalanges), synovitis and carpal tunnel syndrome. Chest radiograph and HRCT are used for screening, diagnosis and monitoring of PSS, and the radiological features include interstitial lung disease (ILD), pulmonary artery hypertension, pericardial effusion, oesophageal strictures or a dilated patulous esophagus (Fig. 2.14.1.2). Gastrointestinal abnormalities include gastroparesis, hypomotility, bacterial overgrowth and malabsorption. Barium meal follow-through examinations can exhibit pseudoobstruction, pneumatosis intestinalis and colonic pseudodiverticula. Systemic lupus erythematosus (SLE) is an autoimmune disease in which immune complex and tissue-binding autoantibody-mediated injury occurs in skin and multiple systemic organs. SLE is more common in women (9:1 female-to-male ratio). A patient is considered to have SLE, if ≥4 of the following ACR criteria are present serially or simultaneously: malar rash, discoid lupus erythematosus lesions, photosensitivity, oral ulcers, nonerosive arthritis involving ≥2 joints, serositis, renal neurological, haematologic and immunologic disorders and positive antinuclear antibody titers. Dermatological manifestations of SLE include lupus dermatitis, rash (malar, discoid), photosensitivity, recurring urticaria, bullae, panniculitis (“lupus profundus”), mucosal ulcers, alopecia, vasculitis, rash and livedo reticularis. Other systemic manifestations include musculoskeletal abnormalities such as polyarthritis, joint deformities of hands and feet, ischaemic necrosis of bone, osteoporosis. Cardiopulmonary features seen on radiograph or CT of chest include pleural and pericardial effusions, lupus pneumonitis, interstitial fibrosis, pulmonary hypertension, pulmonary haemorrhage and the rare pulmonary complication shrinking lung syndrome (Fig. 2.14.1.3). Rarely venous and/or arterial thrombosis may also be seen in SLE. Dermatomyositis (DM) is an inflammatory myopathy with a characteristic cutaneous disease and associated heart, lung and joint involvement. DM is referred to as “overlap syndrome” as it may be associated with other connective tissue diseases (CTDs) such as scleroderma, mixed connective tissue disease (MCTD), Sjögren syndrome, SLE or rheumatoid arthritis. The pathognomonic skin lesions of dermatomyositis are heliotrope rash (violaceous periorbital cutaneous eruptions) and Gottron’s papules (flat, violaceous papules and plaques over bony prominences particularly in the hands). Other dermatological features include midfacial erythema, poikiloderma (combination of telangiectasia, pigmentation and atrophic changes), photosensitivity and alopecia. Systemic manifestations include calcinosis cutis and muscles, vasculitis, pharyngeal or oesophageal muscle involvement, leading to dysphagia, arthritis and interstitial fibrosis of lung (Fig. 2.14.1.4). DM associated with an increased risk of malignancy (6%–60%) includes carcinomas of ovary, breast, lung, nasopharynx, pancreas, colon and nonHodgkin’s lymphoma. Hence, screening for internal malignancy and pulmonary disease is indicated in all adult patients. Rheumatoid arthritis (RA) is the most common chronic systemic inflammatory autoimmune disease characterized by symmetric polyarthritis and many extraarticular manifestations involving the cutaneous, pulmonary, haematologic, cardiovascular and neurologic systems. RA affects nearly 1% of world population and is more frequent in middle-aged females. The characteristic cutaneous manifestations of RA are the rheumatoid nodules, firm, nontender, mobile subcutaneous nodules that occur in about 20%–35% of adult patients. Other dermatological manifestations of RA are palisaded neutrophilic granulomatous dermatitis, interstitial granulomatous dermatitis, nail changes, vascular lesions that include petechiae, purpura, pyoderma gangrenosum, ulcerating plaques and leg ulcers. Radiographs may exhibit the classic musculoskeletal manifestations of symmetric monoarticular/oligoarticular (≤4 joints) or polyarticular (>5 joints) arthritis, joint deformities, tenosynovitis and osteoporosis. Chest radiograph and HRCT can detect pleural effusions, pulmonary nodules, ILD, pulmonary vasculitis and organizing pneumonia (Fig. 2.14.1.5). Early detection of RA may be difficult, and the ACR diagnostic criteria are based on clinical, radiographic and laboratory findings, including the serum rheumatoid factor. Ankylosing spondylitis (AS) and axial spondyloarthritis belong to a group of overlapping disorders which share etiopathological and clinical features and a strong genetic association (HLA B27). Ankylosing spondylitis primarily affects the axial skeleton especially the sacroiliac joints with frequent involvement of peripheral joints and extraarticular structures. Psoriasis is a common skin disease and about one-third of the people with psoriasis develop arthritis (Fig. 2.14.1.6). Comorbidities associated with psoriasis include diabetes, hypertension, dyslipidemia, obesity, metabolic syndrome, psychiatric disturbances, lymphoma, autoimmune disorders and renal disease, and these patients are at higher risk for myocardial infarction, stroke and cardiovascular death. Reactive arthritis refers to acute nonpurulent arthritis complicating an infection elsewhere in the body. SAPHO syndrome (synovitis, acne, pustulosis, hyperostosis, osteitis) is a rare chronic inflammatory disorder involving bone, joint and skin. The common systemic and dermatological features of axial spondyloarthritis are given in Table 2.14.1.2. Systemic-onset juvenile idiopathic arthritis (JIA, Still disease) seen in children and young adults is characterized by polyarthritis, fever, lymphadenopathy, serositis, hepatosplenomegaly and transient cutaneous eruptions of erythematosus macules and papules. Sjögren’s syndrome is a chronic, slowly progressing autoimmune disease characterized by lymphocytic infiltration of the exocrine glands, resulting in xerostomia and dry eyes (keratoconjunctivitis sicca) along with xerosis or dry skin. Polychondritis is a rare disease of cartilage inflammation, which frequently involves ears, nose and laryngotracheal cartilage. Patients may have cutaneous rash, erythematous macules, papules and plaques, subcutaneous calcium deposits and pruritus apart from systemic features such as arthritis, vasculitis, dysphagia, dyspnoea, ILD and an increased risk for malignancy. Sarcoidosis is an inflammatory multisystemic disease of unknown aetiology characterized by the presence of noncaseating granulomas. The dermatological manifestations include erythema nodosum, maculopapular lesions, subcutaneous nodules, plaques, hyper/hypopigmentation and indurated plaque-type lesions involving the face, known as lupus pernio. Granulomatous involvement of bone, muscle, eye, cranial nerves, lung, lymph nodes, heart, liver, kidneys, spleen and rarely, involvement of the breast, testes, ovary or stomach may be seen. Intrathoracic lymphadenopathy and pulmonary manifestations are the most common systemic manifestations of sarcoidosis, and an increased risk of lymphoma is seen in sarcoidosis (Fig. 2.14.1.7). Vasculitis syndromes are a heterogeneous group of diseases characterized by inflammation of and damage to blood vessels affecting either a single organ or several organ systems, leading to ischaemia of the tissues supplied by the involved vessels. Vasculitis can be primary or idiopathic and secondary associated with infections, drugs, malignancies, connective tissue and autoimmune diseases. The classification of vasculitis and the cutaneous manifestations depend on the type and size of the involved vessels. Large vessels are involved in Takayasu arteritis and giant cell arteritis, and cutaneous symptoms include purpura, erythematous subcutaneous nodules, ulcers and pyoderma gangrenosum-like lesions on the extremities. These lesions may be seen at frontotemporal scalp in giant cell arteritis (temporal arteritis or Horton’s arteritis) along with ischaemic optic neuropathy and visual loss, as the disease often affects the extracranial branches of the carotid arteries. The cutaneous findings in medium-vessel vasculitis diseases, polyarteritis nodosa and Kawasaki disease include subcutaneous nodules, ulcers, livedo reticularis, digital infarctions, and papulonecrotic lesions. Similar cutaneous findings are also seen in mixed (medium and small)-vessel vasculitis syndromes such as microscopic polyangiitis, granulomatosis with polyangiitis (Wegener granulomatosis) and eosinophilic granulomatosis with polyangiitis (Churg–Strauss syndrome). Cutaneous small vessel diseases such as IgA vasculitis (Henoch–Schönlein purpura) and cryoglobulinemic vasculitis involve the capillaries, postcapillary venules and nonmuscular arterioles present within the superficial papillary dermis and present with palpable purpura, petechiae, maculopapular rash, vesicles and pustules. Behcet’s syndrome occurs due to systemic perivasculitis and vasculitis of vasa vasorum of large arteries and veins and is characterized by the presence of recurrent oral, genital ulceration, ocular and skin lesions (Fig. 2.14.1.8). The common extracutaneous manifestations associated with cutaneous vasculitis syndromes include arthralgia/arthritis (50%), renal (38%), gastrointestinal (14%), lung (<10%), eye and nervous system (<5%). Many systemic diseases may present with similar cutaneous findings, and hence, a skin biopsy is mandatory to confirm the presence of cutaneous vasculitis. Viral infections such as measles, rubella, varicella and Epstein–Barr virus produce a variety of cutaneous changes, including petechial rashes, maculopapular rashes and vesicular eruptions. Primary HIV infection commonly presents maculopapular rash commonly involving the upper part of the body but can spread to the palms and soles. Herpes zoster presents with grouped vesicles, ulcers and crust in a dermatomal distribution and can lead to secondary infection, scarring and postherpetic neuralgia. Cutaneous changes in bacterial diseases are secondary to bacterial toxins, hypersensitivity reactions, or from direct cutaneous spread of organisms. The clinical manifestations of acute rheumatic fever due streptococcal infection include erythema marginatum, subcutaneous nodules, polyarthritis, carditis and chorea. Rickettsial diseases often present with a cutaneous eruption, fever and headache. Skin examination is very important in the evaluation of patients as several infections present with cutaneous manifestations and the characteristic lesions or distribution of rashes help in narrowing the differential diagnosis of an infection. Disseminated fungal infections such as candidiasis, aspergillosis and mucormycosis are seen in severely immunocompromised patients affecting multiple organs including skin. A wide variety of cutaneous findings linked to internal malignancies are seen associated with paraneoplastic dermatoses, hormone-secreting tumours and inherited syndromes (Table 2.14.1.3). A skin condition is considered as paraneoplastic dermatoses when it is associated with an internal malignancy in the absence of neoplastic cells in skin. It is presumed to occur due to the presence of inflammatory, proliferative or metabolic factors such as hormones, polypeptides, growth factors and cytokines associated with malignancies. The presence of the following five diagnostic criteria, known as Curth’s postulates, establishes the relationship of paraneoplastic dermatoses with internal malignancy: concurrent onset, parallel course, uniform site or cell type of neoplasm, strong statistical and genetic association. Therefore, patients with cutaneous symptoms of diseases that fit Curth’s postulates require mandatory investigations and radiological screening to detect associated neoplasms. Acanthosis nigricans is the most common and the first recognized paraneoplastic dermatosis associated with malignancy and acrokeratosis paraneoplastica (Bazex syndrome) is always associated with an underlying malignancy (Fig. 2.14.1.9). Several conditions such as dermatomyositis, scleroderma, erythroderma, extramammary Paget’s disease, porphyrias have statistically significant association with malignancies and may need further evaluation to rule out them. The sudden unexplained appearance of cutaneous lesions such as necrobiotic xanthogranulomas, vasculitis, pyoderma gangrenosum, vitiligo, multiple seborrheic keratoses (sign of Leser–Trélat), generalized pruritus also seem to have possible association with malignancies. Recurrent and migratory thrombophlebitis (Trousseau’s syndrome) is associated with a risk of an underlying occult malignancy, most frequently of the pancreas, stomach or lung. The cutaneous manifestations of paraneoplastic dermatoses and inherited syndromes that are almost always associated with internal malignancies are given in Table 2.14.1.3. Apart from inherited syndromes, several other malignancies can involve multiple systems including skin. Langerhans cell histiocytosis is a malignancy arising from the dendritic cells (Langerhans cells) of mucocutaneous origin affecting bones, lymph nodes, skin and lung in infants. In adults, variant of multiple myeloma with sclerotic bone lesions can present with POEMS syndrome, an acronym for polyneuropathy, organomegaly, endocrinopathy, M-protein (monoclonal plasma cell dyscrasia) and skin changes which include hyperpigmentation, hypertrichosis, acrocyanosis, flushing and white nails. Recognition of the cutaneous signs and symptoms associated with internal malignancies warrants extensive radiological screening including a CT scan of the chest and abdomen and even MRI or PET-CT to allow early diagnosis of underlying neoplasms, leading to either a complete cure or better prognosis. Several multisystemic diseases involve both the heart and the skin. Patients with antiphospholipid syndrome (APS) may present with deep vein thrombosis, pulmonary embolism, cerebrovascular accidents and recurrent fetal loss. The skin lesions in APS comprising of leg ulcerations, purpura, livedo racemose, vasculopathy and superficial thrombophlebitis may provide a hint to the diagnosis. Hyperlipidemias are associated with coronary artery disease and cutaneous xanthomas. Increased incidence of atherosclerosis, arterial and venous thrombosis along with livedo reticularis, malar rash and diffuse pigmentation abnormalities of skin is seen in homocystinuria. Primary systemic amyloidosis can present as purpura, papules, haemorrhagic bullae of skin and the associated cardiac abnormalities include congestive heart failure, conduction disturbances and cardiomegaly. Ehlers–Danlos syndrome and Marfan syndrome exhibit skin fragility and abnormalities of aorta and mitral valves. Carney complex is associated with cutaneous myxomas, lentigines and atrial myxomas with an increased incidence of endocrine neoplasms (adrenal, pituitary and/or testes). Multiple lentigines and café-au-lait macules are seen in both LEOPARD syndrome associated with hypertrophic cardiomyopathy and Noonan syndrome associated with pulmonic stenosis. Cutaneous features may provide an early diagnostic clue in several conditions such as hemochromatosis and carcinoid syndrome. Gastrointestinal haemorrhage may be associated with hereditary hemorrhagic telangiectasia, pseudoxanthoma elasticum, blue rubber bleb nevus syndrome, Ehlers–Danlos syndrome and vasculitis. The underlying pathology in these conditions includes vascular malformations, arterial rupture, mucosal ulceration and intestinal perforation. Several mucocutaneous bullous and ulcerative diseases such as pemphigus vulgaris, bullous pemphigoid, Behcet’s syndrome and epidermolysis bullosa can affect the oropharynx and oesophagus, particularly the proximal oesophagus with blisters, bullae, webs and strictures. Hereditary GI polyposis syndromes, familial adenomatous polyposis (FAP) and its variant Gardner’s syndrome are characterized by adenomatous GI polyps which invariably undergo malignant transformation and develop into colorectal cancers. Extraintestinal cutaneous lesions in these patients include multiple subcutaneous fibromas, desmoids, lipomas, epidermoid cysts and osteomas, which may precede the GI polyps and serve as initiators for radiological or colonoscopic screening for early removal. Inherited GI hamartomatous polyposis syndromes, Peutz–Jeghers syndrome, Cowden’s syndrome and Cronkhite–Canada syndrome are characterized by the presence of multiple hamartomatous GI polyps of low malignant potential along with cutaneous pigmentation. The characteristic cutaneous features are perioral mucocutaneous pigmentation in Peutz–Jeghers syndrome and hamartomas of skin and mucous membranes, trichilemmomas, mucosal papillomas, lipomas, haemangiomas and neuromas in Cowden’s syndrome. Increased risk of breast, ovary, pancreas and colon cancers is seen in these conditions; hence, patients need surveillance. Acrodermatitis enteropathica and dermatitis herpetiformis (grouped erythematous papules and vesicles over the extensor surfaces) are associated with malabsorption syndromes and celiac disease. Careful evaluation is needed in patients with celiac disease due to the increased incidence of both gastrointestinal and nongastrointestinal neoplasms including bowel lymphoma. The two main types of inflammatory bowel disease (IBD) are ulcerative colitis and Crohn’s disease, in which the skin lesions, pyoderma gangrenosum and erythema nodosum develop after the onset of bowel symptoms along with concomitant active peripheral arthritis (Fig. 2.14.1.10). Several skin and nail changes are noted in patients with hepatic disease. Cirrhosis of liver is associated with spider angiomas, telangiectases, palmar erythema, dilated abdominal wall veins, pruritus and jaundice. The cutaneous findings of pruritus, hyperpigmentation, jaundice and xanthomas are highly suggestive of primary biliary cirrhosis. Disorder of iron metabolism with abnormal deposition of iron in skin, liver, heart, pancreas and endocrine organs is seen in hemochromatosis resulting metallic gray or brown hyperpigmentation of skin. Viral hepatitis may be associated with cutaneous vasculitis, nodules, livedo reticularis, urticaria and pruritus. Cutaneous manifestations secondary to pancreatitis and pancreatic carcinomas include purpura, panniculitis, thrombophlebitis, subcutaneous nodules and necrolytic migratory erythema. Chronic kidney disease (CKD) is associated with changes in skin and nails, alopecia and pruritus. Ultrasound of abdomen in these patients may show bilateral small contracted kidneys. End-stage renal disease (ESRD) or uraemia is also associated with calcinosis cutis in which papules, nodules or plaques may exude a chalky discharge through the epidermis. Calciphylaxis is unique to advanced CKD in which extensive vascular and soft-tissue calcification occurs in small vessels of dermis and subcutaneous tissue, leading to tender, retiform purpuric plaques that frequently ulcerate due to ischaemic necrosis. Nephrogenic systemic fibrosis (NSF) is a rare systemic fibrosing disease occurring in patients with CKD exposed to magnetic resonance contrast agent gadolinium. It is characterized by deposition of collagen in the skin and other organs, hyperpigmented, brawny cutaneous plaques and papules progressing to subcutaneous thickening, induration and debilitating joint contractures. Current recommendations are that patients with CKD stage 3 (GFR 30–59 mL/min) should minimize exposure to gadolinium, and those with CKD stages 4–5 (GFR <30 mL/min) should avoid the use of gadolinium agents unless it is medically necessary. Renal transplant recipients are at increased risk for cutaneous infections and cutaneous malignancies secondary to immunosuppression. Endocrine diseases occur due to three conditions: hormone deficiency, hormone resistance and excessive production of hormones. Many of these disorders have concurrent mucocutaneous manifestations that can provide clues for early diagnosis. Pituitary gland tumours are a part of several familial syndromes. Pituitary adenomas and skin lesions are seen in multiple endocrine neoplasia syndromes. Carney complex is characterized by spotty skin pigmentation (lentigines) and pituitary hyperplasia or adenoma. Hypersecretion of growth hormone by pituitary hyperplasia or tumour can cause acromegaly. Acromegaly with skin changes is seen in McCune–Albright syndrome and familial acromegaly syndromes. Cushing’s disease referring specifically to Cushing’s syndrome caused by a pituitary corticotrope adenoma is characterized by thin skin, purple striae, easy bruisability, hirsutism and acne. Skin changes may be the first clue to the diagnosis of hypopituitarism which can manifest with thin skin, scant body hair and pale complexion. Pituitary adenoma is the commonest cause of hypopituitarism and hyperpituitarism, and MRI of brain is indicated in suspected cases for the detection of both micro- and macroadenomas. Thyroid gland abnormalities such as Hashimoto’s thyroiditis, multinodular goiter or thyroid carcinoma may present with skin, hair and nail changes. Thyroid malignancies are seen in multiple endocrine neoplasias (MEN syndromes), Cowden’s disease, Gardner’s syndrome and Carney’s complex, and the cutaneous manifestations in these syndromes include mucosal neuromas, neurofibromas, diffuse lentigines and café-au-lait macules. The cutaneous features of hypothyroidism are dry, coarse, cold skin, myxedema and alopecia, whereas hyperthyroidism is associated with warm and moist skin, hyperpigmentation, pruritus, pretibial myxedema and thyroid dermopathy/acropachy. Ultrasound is useful in the investigation of focal nodules associated with thyroid hormone abnormalities. Disorders of the adrenal cortex can present with both over- and underproduction of adrenal hormones, leading to characteristic skin findings. Addison’s disease or primary adrenocortical insufficiency can present with hyperpigmentation of the skin and mucous membranes with prominent involvement of sun-exposed skin, sites of trauma and skin creases. Cushing disease occurs due to chronic exposure to excess glucocorticoids characterized by fragile skin, thin epidermis, abdominal striae, purpura, poor wound healing, cutaneous infections, acne, hirsutism and altered subcutaneous fat distribution (moon facies, buffalo hump). Excessive androgens seen in congenital adrenal hyperplasia and polycystic ovary syndromes can result in precocious puberty in children and virilization, hirsutism, acne and male-pattern alopecia in adults. Pheochromocytomas and paragangliomas are catecholamine-producing tumours derived from the sympathetic or parasympathetic nervous system. These tumours can occur sporadically or as a part of inherited syndromes (MEN syndrome and von Hippel– Lindau (VHL) disease). Pheochromocytomas producing excessive epinephrine can cause paroxysmal flushing of face, chest and upper extremities. Plain and contrast-enhanced CT and MRI of abdomen are indicated in suspected patients for the diagnosis of adrenal lesions and for differentiating benign from malignant lesions. Diabetes mellitus is a highly prevalent, chronic disease affecting multiple organs including skin. The cutaneous manifestations include acanthosis nigricans, diabetic bullae, diabetic dermopathy, necrobiosis lipoidica (yellow atrophic skin patches) and neuropathic leg ulcers. Diabetic foot is characterized by the presence of peripheral neuropathy and peripheral vascular disease, leading to ulceration over the bony prominences of the foot and ankle, which can be further complicated by secondary infection, gangrene and Charcot arthropathy. Cutaneous xanthomas seen as yellowish papules, nodules and plaques can indicate the presence of underlying metabolic syndromes and hyperlipidemias. Multisystemic disorders which can have both pulmonary and cutaneous manifestations include genetic syndromes (Birt–Hogg–Dubé syndrome and tuberous sclerosis), autoimmune/inflammatory (granulomatosis with polyangiitis, eosinophilic granulomatosis with polyangiitis, dermatomyositis and systemic sclerosis), infections (tuberculosis, cryptococcosis, aspergillosis and varicella) and neoplasms (Kaposi sarcoma, paraneoplastic pemphigus associated with lymphomas). Congenital hereditary haemorrhagic telangiectasia (Osler–Weber–Rendu disease) is a rare genetic disorder associated with mucosal and cutaneous telangiectasias and arteriovenous malformations in lungs, brain and liver. Sarcoidosis is a multisystem granulomatous disease of unknown aetiology with several cutaneous and pulmonary manifestations. Intrathoracic diseases, including hilar adenopathy and pulmonary parenchymal disease, are seen in more than 90% of cases with sarcoidosis. The staging of pulmonary sarcoidosis (0-IV) is based on chest radiographic findings. The initial pulmonary findings on HRCT include micronodules, macronodules, alveolar and ground-glass opacities. These early changes occurring due to granulomatous inflammation are reversible, but later the disease progresses to chronic irreversible fibrosis and HRCT may show traction bronchiectasis, architectural distortion, volume loss, interlobular septal thickening and honeycombing. The genetic disorders characterized by cutaneous lesions along with an increased risk of brain tumours were grouped together as neurocutaneous syndromes and the important of these are neurofibromatosis (NF or Von Recklinghausen disease), tuberous sclerosis (TS or Bourneville disease), Sturge–Weber syndrome (SWS) and von Hippel–Lindau syndrome (VHL). Except for VHL, these syndromes have characteristic cutaneous manifestations, some of which are included in the diagnostic criteria. Patients with NF1 may have central nervous system (CNS) tumours including neurofibromas, optic nerve gliomas, astrocytomas and meningiomas (Fig. 2.14.1.11). The other systemic manifestations of NF1 include with hamartomas of the iris termed Lisch nodules, pheochromocytomas, pseudoarthrosis of the tibia, scoliosis, epilepsy and mental retardation. The cutaneous manifestations of NF1 include café-au-lait spots (flat brown macules) and axillary freckling. Patients with tuberous sclerosis may have seizures, mental retardation, renal angiomyolipomas, cardiac rhabdomyomas and the CNS lesions including subependymal nodules, cortical tubers and subependymal giant cell astrocytomas (SEGAs). The characteristic cutaneous lesions of TS are adenoma sebaceum (facial angiofibromas), shagreen patches, hypomelanotic macules (ash leaf macules) and periungual fibromas (Koenen’s tumours) (Fig. 2.14.1.12). Sturge–Weber syndrome is characterized by a facial capillary malformation known as port-wine stain on the forehead and upper eyelid, in the area of the ophthalmic branch of the trigeminal nerve, in association with leptomeningeal angiomatosis and glaucoma (Fig. 2.14.1.13). Cutaneous port-wine stain may also be seen in Cobb syndrome and VHL, though the incidence of cutaneous abnormalities is very less in VHL.
2.14: Radiological imaging of skin and subcutaneous disorders
Immune-mediated, inflammatory and rheumatologic disorders
Other autoinflammatory systemic diseases
Infectious diseases
Cutaneous manifestations of internal malignancy
Diseases/Syndromes
Cutaneous Findings
Internal Malignancy
PARANEOPLASTIC DERMATOSES
Acanthosis nigricans
Velvety hyperpigmentation of flexural surfaces
Gastrointestinal (GI) and genitourinary (GU) adenocarcinomas
Acrokeratosis paraneoplastica (Bazex syndrome)
Erythematous/violaceous, macules progressing to psoriasiform plaques in acral regions, nail ridging
Squamous cell carcinomas of the upper aerodigestive tract, solid malignancies and lymphomas
Paraneoplastic pemphigus
Erythematous papules, vesicles, bullous and lichenoid lesions
Lymphoproliferative malignancies and thymoma
Acquired hypertrichosis lanuginosa (malignant down)
Appearance of fine lanugo hairs
Lung, colon and other internal malignancies
Erythema gyratum repens
Erythematous gyrate or serpiginous lesions
Lung (most common), breast, GU and GI neoplasms
Acute febrile neutrophilic dermatoses (Sweet syndrome)
Pyoderma gangrenosum
Acute myelogenous leukaemia, myelodysplasia, plasma cell dyscrasias
HORMONE SECRETING SYNDROMES
Carcinoid syndrome
Flushing, erythema, pellagra-like dermatitis and sclerodermoid changes
Bronchial and GI carcinoid tumours, hepatic carcinoid metastases
Ectopic ACTH syndrome
Features of Cushing’s syndrome, hyperpigmentation
Lung (most common), GI carcinomas
Glucagonoma syndrome
Necrolytic migratory erythema
Pancreatic carcinoma
INHERITED SYNDROMES
Ataxia telangiectasia
Telangiectasia
Breast, leukaemia, lymphoma
Cowden’s syndrome
Facial trichilemmomas, keratotic papules
Breast, thyroid and GI carcinomas
Familial adenomatous polyposis
Benign skin tumours such as lipomas, fibromas and epidermal cysts
GI malignancies
Muir–Torre syndrome
Sebaceous tumours, keratoacanthomas
GI and GU carcinomas
Peutz–Jeghers syndrome
Mucocutaneous (perioral) pigmented macules
Breast, pancreatic, ovarian, testicular and cervical cancers
Multiple endocrine neoplasia syndromes
Facial angiofibromas, collagenomas, melanosis guttaca, lipomas, melanomas, “café-au-lait” spots (flat brown macules)
Pituitary, parathyroid, pancreas, pheochromocytoma and medullary thyroid carcinomas
Neurofibromatosis
Café-au-lait spots, freckling, cutaneous neurofibromas
Nerve sheath tumours, brain tumours, rhabdomyosarcoma, pheochromocytomas
Gorlin’s syndrome
Nevi, basal cell carcinoma
Basal cell carcinoma, medulloblastoma, jaw cysts
Tuberous sclerosis
Hypomelanotic macules (ash-leaf spots), shagreen patches, angiofibromas
Renal neoplasms
Birt–Hogg–Dubé syndrome
Triad of fibrofolliculoma, trichodiscoma, and acrochordon.
Renal neoplasms, hereditary leiomyomatosis
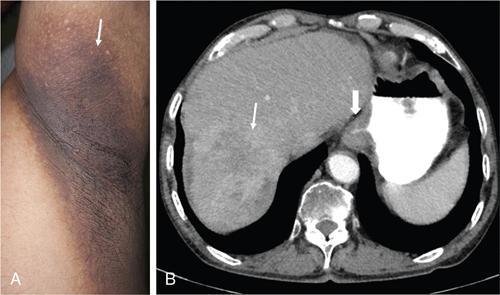
Cardiovascular diseases
Gastrointestinal disorders
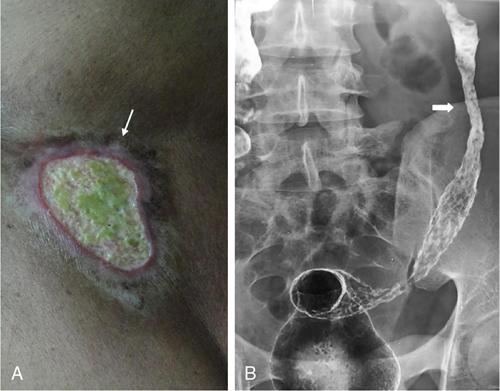
Renal disorders
Endocrine and metabolic diseases
Pulmonary diseases
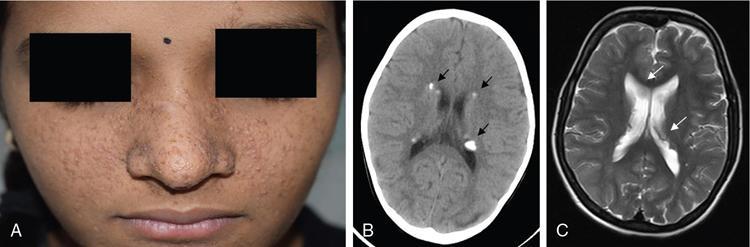
Neurocutaneous syndromes (phakomatoses)
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


