and Ashutosh Dash2
(1)
Nuclear Security and Isotope Division, Oak Ridge National Laboratory, OAK RIDGE, USA
(2)
Isotope Production and Applications Division, Bhabha Atomic Research Centre, Mumbai, India
7.1 Introduction
Radionuclide generators represent important in-house production systems which can provide daughter radioisotopes generated by parent decay on-demand without the need for local access to an accelerator or research reactor. The 99Mo/99mTc generator represents the most important example and provides NCA 99mTc without which the success of diagnostic nuclear medicine could not have been possible. It is estimated that 99mTc is used in over 30 million studies annually on an international basis. The in-house or central radiopharmacy use of radionuclide generators has many advantages, since their use represents a cost-effective strategy for obtaining the desired daughter radionuclide on demand and also ensures availability of the daughter radionuclides in high specific activity and usually in no-carrier-added form. The most widely used technology for fabrication of radionuclide generators for clinical applications is the column chromatography system. However, in recent years enormous interest has developed for use of other novel separation techniques to provide therapeutic radionuclides. Among many other requirements, such as those relating to establishment of chemical, radiochemical, and radionuclidic purity of the daughter radionuclide, radionuclide generators intended for clinical applications must be manufactured and operated under conditions of good manufacturing practice (GMP) guidelines since these products represent active pharmaceutical ingredients (APIs) which will be incorporated into radiopharmaceutical products prepared for human use.
In addition to a variety of other radionuclide generator systems which provide diagnostic radioisotopes (Osso and Knapp 2011; Roesch and Knapp 2011; Knapp and Mirzaeh 1994; Knapp and Butler 1984; Mirzadeh and Knapp 1996; Chakravarty and Dash 2013; Knapp et al. 2014), radionuclide generator systems also represent important resources to provide a variety of therapeutic radioisotopes for applications in nuclear medicine, oncology, and interventional specialties (Table 7.1). Many parent–daughter pairs have been evaluated as radionuclide generator systems (Roesch and Knapp 2011; Dash and Chakravarty 2014a, b; Knapp et al. 2014; Pillai et al. 2012). While only a few therapeutic generators are currently used in routine clinical practice, several others show great promise for further development and potential widespread clinical use. An important requirement for radionuclide generator systems for routine clinical use is a long half-life of the parent radionuclide, which provides logistics for production, transportation, and reduced injection dose costs because of long useful shelf lives. A variety of radionuclide generators which provide daughter radionuclides which are used and have been proposed for therapeutic applications are summarized in Table 7.1. The most widely used radionuclide generators for therapeutic applications include β-emitting daughters from the 188W/188Re and 90Sr/90Y generator systems and alpha-emitting radioisotopes obtained from the 225Ac/213Bi and 227Ac/223Ra generator systems.
Table 7.1
Key examples of radionuclide generators providing therapeutic radioisotopes
Generator system | Main production route of parent radionuclide | Parent radionuclide | Daughter radionuclide | ||
|---|---|---|---|---|---|
T ½ | Main decay mode | T ½ | Main decay mode | ||
225Ac/213Bi | Decay chain | 10.0 days | α | 45.6 min | β−, α |
227Ac/223Ra | Reactor decay chain | 21.7 years | β−, then α through 227Th | 11.4 days | α |
166Dy/166Ho | Reactor | 3.40 days | β− | 1.12 days | β− |
194Os/194Ir | Reactor | 6.0 years | β− | 19.15 h | γ, β− |
212Pb/212Bi | 228Th (T ½ = 1.913 a) through 224Ra | 10.4 h | β− | 60.55 months | α |
226Ra/222Rn | Decay chain | 1.6 × 103 years | α | 3.83 days | α |
90Sr/90Y | Reactor (fission product) | 28.5 years | β− | 2.67 days | β− |
188W/188Re | Reactor | 69.4 days | β− | 16.98 h | β− |
Radionuclide generators provide therapeutic radionuclides which can decay by emission of beta particles, Auger electrons, low-energy photons, and alpha particles and are thus useful for a variety of therapeutic applications in nuclear medicine, oncology, and other clinical specialties. During the last decade, there has been a tremendous increase in the development and use of new therapeutic radiopharmaceuticals radiolabeled with radionuclides which are available from radionuclide generator systems (Pillai et al. 2012; Knapp et al. 2014; Chakravarty et al. 2008; Miederer et al. 2008; Scheinberg and McDevitt 2011; Zalutsky and Pozzi 2004). The availability of generator-derived therapeutic radionuclides is necessary for the development and testing and commercialization of agents with potential for endoradiotherapy (ERT) and has stimulated the development of an increasing spectrum of therapeutic tracers. Interest and the importance of the availability of therapeutic radionuclides available from generator systems have developed from the availability of several fully automated operations of many generators, coupled with parallel success in the development of therapeutic targeting agents, such as antibodies, peptides, and other agents. Applications of these generator-derived therapeutic radioisotopes include cancer therapy, synovectomy for the treatment of synovitis, and more recently therapy of skin cancer and inhibition of arterial restenosis after angioplasty of the coronary and peripheral vessels. A key example is the recent approval by the US Food and Drug Administration (FDA) for the use of the 90Y-labeled murine anti-CD20 antibody “Zevalin” (ibritumomab tiuxetan) for the treatment of patients with low-grade, follicular or transformed non-Hodgkin’s lymphoma. This agent is the first antibody radiolabeled with a generator-derived therapeutic radionuclide and represents the first of a variety of expected new therapeutic radiopharmaceuticals for oncologic applications.
7.2 Production of Parent Radionuclides
The production of the parent radioisotopes required for generator fabrication is an important issue. Production requires the availability of research reactors and various types of accelerators, as described in Chap. 5 and 6. Most therapeutic radionuclides currently in clinical use are neutron rich and characterized by beta decay and available from reactor production (Chap. 5), since neutron capture by the target nuclide forms a radioactive or unstable product which decays by beta emission. Key examples of therapeutic radionuclides obtained from reactor-produced parent radionuclides include holmium-166 (166Ho), from the dysprosium-166 (166Dy)/166Ho generator, and rhenium-188 (188Re), from the tungsten-188 (188W)/188Re generator. Several generator parent radionuclides are produced during nuclear fission and include 90Sr, isolated from fission products as the parent for the 90Sr/90Y generator. Recovery of radioactive parents from “extinct” radioactive decay processes represents another strategy to obtain generator parents. Thorium-229 (T ½ = 7340 years) is an important source to obtain 225Ac as the parent of the 225Ac/213Bi generator system. The 211At α-emitter is also of interest for targeted alpha therapy and can be obtained by the decay of 14.6 h 211Rn (Vachtel et al. 1976; Visser et al. 1979; Mirzadeh and Lambrecht 1980), although usually accelerator-produced by the 209Bi(α,2n) route.
7.3 Decay and In-Growth Principles
The relationships between parent radioisotope decay, daughter in-growth, and timing of daughter elution have been widely discussed and described in the literature (Roesch and Knapp 2011; Osso and Knapp 2011). The decay and growth of radionuclides are described by exponential equations which were originally formulated by Rutherford and Soddy (Rutherford and Soddy 1902) and the widely used Bateman equations (Bateman 1910) which were later developed to relate decay and in-growth of the naturally occurring actinium, uranium, and thorium series. As described in the following equations, at any time t, the activity A of a radioisotope is related to the number of atoms N, where λ represents the radionuclide decay constant.
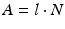
The radionuclide decay with time follows the exponential law:
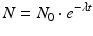
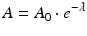
where N and A represent the number of atoms and activity at time t and N 0 and A 0 the quantities when t = 0, respectively. The time required to reduce the activity of the radionuclide to 50 % of its initial value is the half-life, T 1/2. This value is related to λ by the following equation:
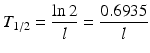
The daughter decay product radioisotope is generally denoted by subscript 2 and the parent radioisotope by subscript 1. The following equations then relate the decay of the parent and in-growth and decay of the daughter:
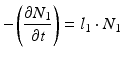
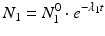
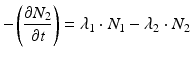
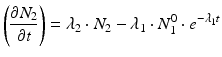
Solving the equations above and assuming that when t = 0, 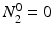 the equation can then be solved as
the equation can then be solved as
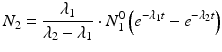
This equation can be simplified to
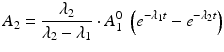
(7.1)
(7.2)
(7.3)
(7.4)
(7.5)
(7.6)
(7.7)
(7.8)
the equation can then be solved as(7.9)
(7.10)
Use of this equation permits calculation of the daughter activity level at any time t when the initial parent activity level is known. This equation is used to estimate the daughter activity in-growth curves illustrated for a hypothetical parent–daughter pair in Fig. 7.1, where successive elutions and in-growth of the daughter radioisotope are illustrated.
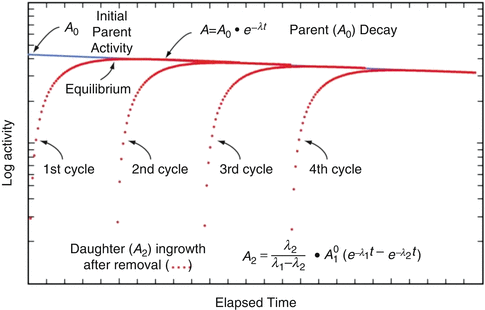
Fig. 7.1
Illustration of parent–daughter decay, daughter in-growth, and cycling of daughter by successive generator elutions (From Osso and Knapp 2011)
There are three types of equilibria which can arise which are dependent on the relative parent–daughter physical half-lives. In the case of “transient equilibria,” the parent half-live is longer than the daughter half-life, i.e., ![$$ {T}_{1/2(1)>}{T}_{1/2(2)} $$
” src=”/wp-content/uploads/2017/04/A339591_1_En_7_Chapter_IEq2.gif”></SPAN> or <SPAN class=EmphasisTypeItalic>λ</SPAN> <SUB>1</SUB> < <SPAN class=EmphasisTypeItalic>λ</SPAN> <SUB>2</SUB>. Equation (<SPAN class=InternalRef><A href=]() 7.9) can be simplified when
7.9) can be simplified when 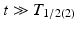 , i.e., the decay time is much longer than the daughter half-life:
, i.e., the decay time is much longer than the daughter half-life:
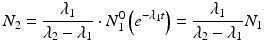
which can also be expressed as
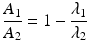
, i.e., the decay time is much longer than the daughter half-life:(7.11)
(7.12)
After reaching equilibrium, the daughter decays with the half-life of the parent and the activity of the daughter will be higher than the parent activity. Alternatively, in the case of “secular equilibria,” the parent half-life is much longer than the daughter half-life, and this is quite useful for the generator to serve as an in-house radioisotope production system, where the daughter can be successively repeatedly eluted over a long time period following sufficient in-growth between elutions. In this case, T 1/2(1) > > T 1/2(2) or λ 1 < <λ 2. For a typical radionuclide generator of this type which provided a short-lived therapeutic radioisotope from a radioactive pair at transient equilibrium, the 188W (T 1/2 = 69 days)/188Re (T 1/2 = 16.9 h) generator is an important example discussed in Sect. 7.6.1.2. In this case, when radioactive equilibrium is achieved, Eq. (7.9) can be simplified to
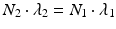
or
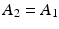
(7.13)
(7.14)
The relative growth and decay of the parent and daughter radionuclide pair is illustrated in Fig. 7.2a (transient equilibrium) and Fig. 7.2b for secular equilibrium.
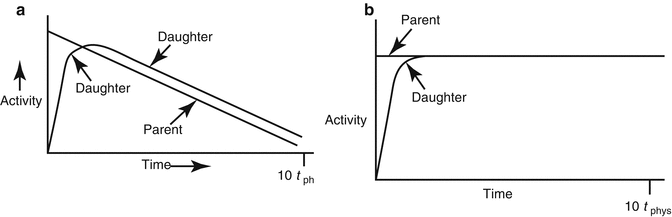
Fig. 7.2
Illustration of secular and transient equilibrium. (a) Transient equilibrium is a condition when the t ½(phys) of the parent is approximately 10 times greater than the t ½(phys) of the daughter. (b) Secular equilibrium is a condition when the t ½(phys) of the parent is many times greater than the t ½(phys) of the daughter(100–1000 times greater or more)
Finally, in some cases, “no equilibrium” is achieved, and the half-life of the parent is shorter than the daughter half-life, i.e., T 1/2(1) < T 1/2(2) or λ 1 > λ 2. This is an impractical situation to provide daughter radionuclides for clinical use, and there is no practical application, since the time (T max) required to reach the maximum daughter activity level is
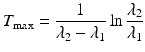
(7.15)
For practical considerations, radionuclide generators are eluted at periodic intervals depending on the daughter activity requirements. Often the separation of the daughter from the parent may not occur at the time the daughter activity is at its maximum. Owing to the decrease of the parent radionuclide activity within the time between two elution steps, the radionuclide generator is expected to provide a reduced level of the daughter radionuclide activity by each subsequent elution as shown in Fig. 7.3.
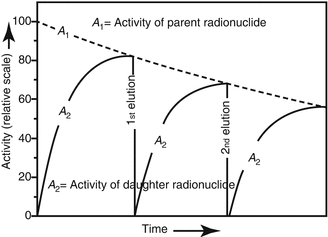
Fig. 7.3
Example of radionuclide daughter activity in-growth over repeated elutions. The radionuclide generator provides a reduced level of the daughter radionuclide activity by each subsequent elution
When the daughter product is long-lived, periodic elution will take place prior to reaching the maximum equilibrium daughter activity levels and it is normal to use generators in this way. As an example, fifty percent daughter activity in-growth is detected in one half-life and 75 % in two half-lives, and the daughter activity reaches the activity of the parent radionuclide in 5–6 half-lives (Fig. 7.4).
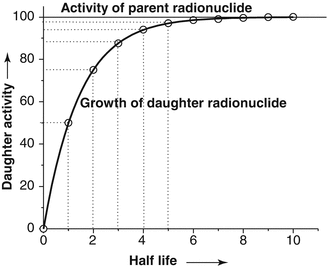
Fig. 7.4
The growth of daughter radionuclide in a pure parent fraction as a function of time. Depending upon the growth of the daughter radionuclide activity in the generator, different levels of the daughter radionuclide activity can be separated
The activity eluted from the generator typically follows a Gaussian distribution as shown in Fig. 7.5, with the maximum activity being eluted in the intermediate fractions. The eluent can also be “fractionated” by discarding those fractions with low activity if the activity concentration is a critical issue.
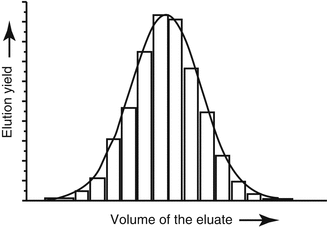
Fig. 7.5
Activity eluted from a generator which generally follows a Gaussian distribution
7.4 Radiochemical Separation of Therapeutic Radionuclides
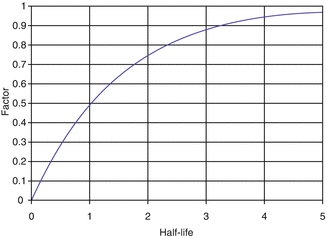
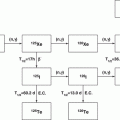
Following reactor or cyclotron irradiation and discharge, the targets require subsequent radiochemical processing to obtain the desired radionuclide in high radionuclidic and radiochemical purity. In general, the radionuclides produced by reactor/accelerator activation are present in only microscopic levels and are diluted with target material and other radionuclidic impurities which are co-produced. In most cases the product radionuclide is chemically distinguishable from the target material, as illustrated in Fig. 7.6. It is necessary for the atomic number Z to change during the transmutation process for the product radionuclide to be available carrier-free from processing the irradiated target. This often occurs during accelerator production (Chap. 6), when a proton is added to the nucleus. In contrast, for most reactor-produced radioisotopes, the nuclear insertion of a neutron does not change the atomic number, resulting in the formation of product radioactive atoms which have the same Z as the target atoms. Since separation by the normally available methods is not possible, the product is diluted by the target atoms and thus is not available NCA. However, specific activity values for reactor-produced radioisotope vary greatly, depending on several factors, which include the neutron capture cross section, and if the product is formed by an alternative route such as beta decay of the initial radioactive product (Mirzadeh and Knapp 1996; Mausner et al. 1998). In addition, although higher epithermal neutron cross-section values are low and permit only low production yields, products formed by the (n,p) route can also be obtained in high specific activity following chemical processing. Since the product radionuclide of interest must be chemically separated from the target as well as from other radioactive and nonradioactive contaminants, however, the availability of appropriate radiochemical separation procedures is essential.
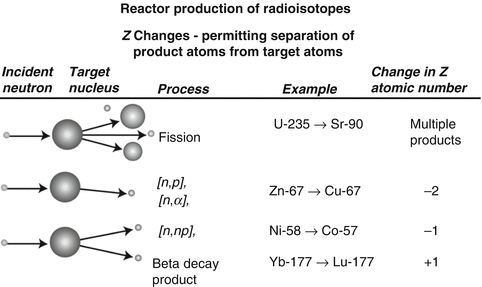
Fig. 7.6
Examples of production pathways from which carrier-free and carrier-added radioactive products can be obtained via reactor production when the Z changes
Radionuclide Production Yields in a cyclotron is given by the equation
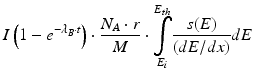 where I = beam current, N = number of atoms, λ A = probability of transforming target atom A into B, and E = energy of the beam.
where I = beam current, N = number of atoms, λ A = probability of transforming target atom A into B, and E = energy of the beam.
The term 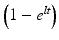 is known as activity build-up factor. Plot of this factor with half-life is shown in Figs. 7.7 and 7.8.
is known as activity build-up factor. Plot of this factor with half-life is shown in Figs. 7.7 and 7.8.
is known as activity build-up factor. Plot of this factor with half-life is shown in Figs. 7.7 and 7.8.Fig. 7.7
Buildup of activity in a cyclotron
Fig. 7.8
Examples of key reactor routes for production of radioisotopes with no change in Z
Conventional chemical separation technologies can generally be modified as required to isolate and concentrate radionuclides of interest. A variety of radiochemical separation procedures are employed to isolate the radionuclide of interest with the required purity. Each separation procedure has its own distinct advantages and disadvantages (Dash and Knapp 2015; Dash and Chakravarty 2014a, b; Dash et al. 2013), but since each radionuclide and its target have different chemistry, many options need to be evaluated. These methods can be adapted either alone or in different combinations with each other, depending upon the chemical nature of the target and radionuclide of interest to be separated with desirable purity and acceptable yield. Radiochemical separation of radionuclides presents a number of basic challenges owing to the often limited radiation stability of the conventional sorbents and solvents, extremely high selectivity and high purity requirements of the radionuclide to be separated. Some of the radiochemical separation procedures employed in radionuclide production are discussed below.
7.5 Methods for Parent–Daughter Separation
The methods available for separation of the parent and daughter chemical species which are amendable and practical for radionuclide generator development (Dash and Knapp 2015; Dash et al. 2013; Dash and Chakravarty 2014a, b) are extensive and include many well-known technologies described in the following sections. Several separation processes summarized in Table 7.2 are based on differences in physical and chemical properties that have been exploited.
Table 7.2
Examples of useful separation process that have been applied for radionuclide separation systems
Separation method | Physical/chemical property for parent–daughter separation | Basis |
|---|---|---|
Column chromatography | Charge | Difference in adsorption on an adsorbent |
Solvent extraction | Hydrophobicity | Difference in solubility in two phases |
Sublimation | Vapor pressure | Difference in vapor pressure |
Precipitation | Solubility | Difference in solubility product |
Solid-phase column extraction | Hydrophobicity | Difference in affinity |
Electrochemical | Standard electrode potential (Eo) | Difference in standard or formal electrode potential |
Extraction chromatography | Specific chemical interaction | Difference in affinity of solutes dissolved in a liquid for an extractant immobilized in solid |
Supported liquid membrane (SLM) | Chemical energy | Difference in solubility on the liquid membrane |
7.5.1 Ion Exchange Column Chromatography
This is the most practical and widely used technology for radionuclide generator separations which relies on the reversible exchange of ions in solution with ions electrostatically bound to an insoluble support media based on their charge density and interaction between the ions and support media. This technology is usually performed in columns in which the insoluble stationary phase generally preferentially interacts with the particular charged ions (radioactive parent) to permit separation from the desired daughter radionuclide species. The radionuclide species uptake properties and chemical selectivity characteristics of a wide variety of ion exchange materials for a range of elements are available in the literature. Using this information the radiochemical separation scheme for a given parent–daughter pair often be rationally designed. Ion exchange column chromatography currently developed has been refined to such a degree that this technology represents a highly selective and efficient technique that can separate closely related radionuclide from radionuclide mixtures. Table 7.1 summarizes the properties of typical radionuclide generators of current interest which provide therapeutic daughters. A schematic diagram of a generator housed in a lead-shielded container to provide the daughter radionuclide is shown in Fig. 7.9.
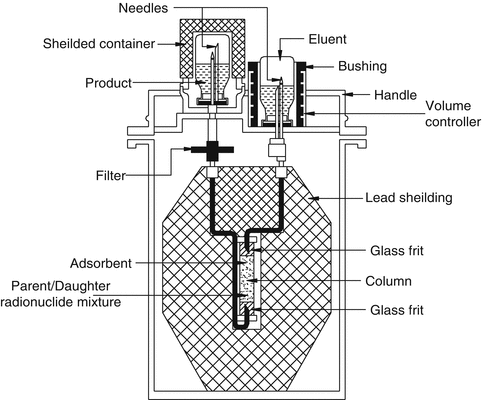
Fig. 7.9
Schematic diagram of a column chromatography generator housed in a lead-shielded container to provide daughter radionuclide. The column containing a bed of adsorbent binds the parent radionuclide and eluent from the inlet side is drawn through the column and into the outlet vial for daughter radionuclide collection
7.5.2 Solvent Extraction
The process of selective transfer of a component in a mixture of two or more components based on the differences in solubility in two immiscible liquid phases is the basis of the solvent extraction technique. In this process two immiscible liquids—usually one aqueous and another organic—are vigorously shaken in an attempt to effectively disperse and efficiently contact with one another. In this manner, the radionuclide of interest which generally has higher organic solubility can migrate from the aqueous phase into the organic phase. Conventional liquid–liquid extraction in the classical version has seldom been used for the radiochemical separation of radionuclides because of the required use of expensive special-grade solvents. In addition, the issue of the difficulties in reaching complete extraction into the organic phase and the generation of organic liquid radioactive wastes are other factors. For the use of solvent extraction for radiochemical separation, extraction with solvent volumes as low as several milliliters and the use of special portable vessels under dynamic conditions are preferred, which places challenges on the widespread, high-activity use of this technology. However, one example in the diagnostic arena is the use of methyl ethyl ketone (MEK) for the effective extraction of NCA 99mTc from low-specific-activity 99Mo (Anwar et al. 1968; Skuridin and Chibisov 2010; Zykov et al. 2001). Schematic diagram of the 99Mo/99mTc MEK extraction generator is depicted in Fig. 7.10.
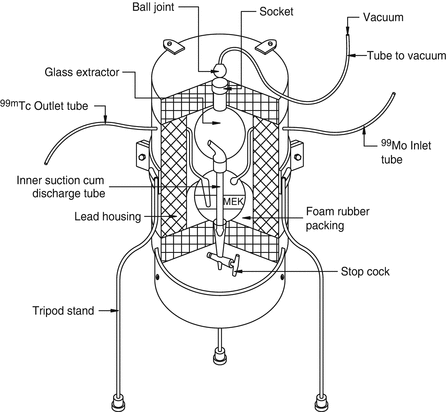
Fig. 7.10
Schematic diagram of the 99Mo/99mTc MEK extraction generator. Methyl ethyl ketone (MEK) is introduced through the inlet tube into the container containing Na2MoO4 in 4M NaOH to extract 99mTc into the organic phase
7.5.3 Distillation
Distillation is a separation technique in which differences in element vapor pressures or their compounds are exploited. This technology essentially takes advantage of the differences in the boiling points of the constituents to separate a desired radionuclide from a mixture. It is a useful separation tool if the radionuclide of interest is volatile or can be transformed into a volatile compound. Separation of radionuclide of interest from others is achieved by vaporization at the boiling point of the mixture and subsequent condensation of the vapor. This technique provides clean separations, provided that proper precautions are taken to avoid contamination of the distillate. Precautions are sometimes necessary to avoid loss of volatile radioactive substances during the dissolving of irradiated targets or during irradiation itself. This technology has been avidly exploited for the separation of 99mTc from 99Mo (Christian et al. 2000; Gerse et al. 1988; Zsinka 1987; Macháň et al. 1981). Operational principle of a 99Mo/99mTc sublimation generator is illustrated in Fig. 7.11.
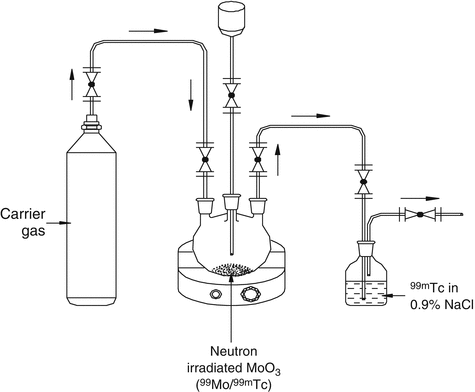
Fig. 7.11
Operational principle of a 99Mo/99mTc sublimation generator. Carrier gas is allowed to pass through neutron-activated target and the separation of 99mTc is performed at sublimation temperature
7.5.4 Precipitation
Precipitation is also a conventional separation technology in which the solubility of metal ions, salts, or complexes is exploited for the selective separation of one metal ion from the other. When the solubility limit of the solute in the solvent exceeds, the material appears as a precipitate and is separated from the other component(s). Effective separation of a radionuclide is accomplished by the judicious selection of the compound that forms the precipitate and the concentration of solutions used in the precipitate formation. Precipitation generally is followed by either centrifugation or filtration for the effective separation of the precipitate. The speed of separation is dictated by the nature of the precipitate (i.e., gelatinous, crystalline, amorphous, etc.), the sample size being processed, and other factors.
7.5.5 Extraction Chromatography
The possibility to incorporate an extractant or a solution of an extractant into an inert substrate is the genesis of the extraction chromatography (EXC). The concept of extraction chromatography separation seemed very attractive as it would exploit the selectivity of liquid–liquid extraction and at the same time offer the ease of operation of column-based separation system. Recent advances in ligand design, in particular the introduction of novel extractants capable of strong and selective complex formation with metal ion, have enhanced the scope of the technique (Dietz and Horwitz 2000; Jassin 2005).
7.5.6 Solid-Phase Column Extraction
An alternative to solvent extraction is the separation of metal ion by direct contact of the aqueous sample with a sorbent usually using column chromatographic technique. Desorption of metal ion of interest can be carried out by elution with a suitable solvent or solvent mixture. Optimal desorption of metal ion from the column is determined by the selectivity (distribution coefficient) of the metal ions in the organic solvent/extractant. Utility of this technique has also tried for the development of 99Mo/99mTc and 72Se/72As radionuclide generators (Braun et al. 1987; Jennewein et al. 2005; Chattopadhyay et al. 2008).
7.5.7 Electrochemical Separation
The strategy to exploit the differences in the standard metal ion reduction potential of is the basis to separate metal ions of interest from other species under the influence of an applied potential difference. A mixture of radionuclides having adequate difference in their formal potential values in an electrolytic medium can be mutually separated by selective electrodeposition of one radionuclide on an electrode surface under the application of the applied potential. Electrochemical strategy has been used for the development of 99Mo/99mTc, 188W/188Re, and 90Sr/90Y generators (Dash and Chakravarty 2014a, b; Chakravarty et al. 2008, 2009, 2010a, b, c, 2012a, b). An electrochemical method based on the different reducibility of lanthanides and the ability to form amalgams has been utilized successively, for instance, for purification of 177Lu (Chakravarty et al. 2010a, b, c). Under suitable conditions of electrolysis, the divalent lanthanide forming more easily the amalgam is preferentially transferred into the mercury electrode from other trivalent lanthanides. The amalgated lanthanide can again be transferred into solution by washing of mercury with dilute acid. Electrochemical separation is very specific and, therefore, can be successfully used in the presence of many other radionuclides. Schematic representation of electrochemical separation strategy is depicted in Fig. 7.12.
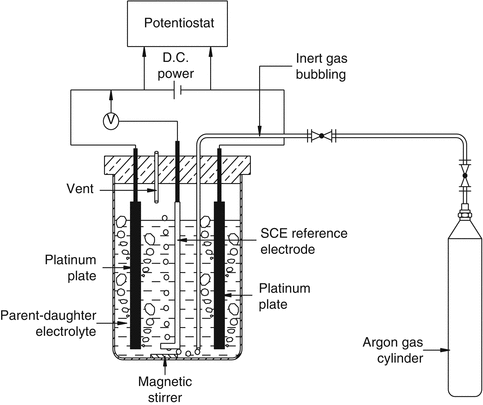
Fig. 7.12
Electrochemical separation strategy. Separation is primarily based on the selective electrodeposition of daughter radionuclide on a platinum cathode surface from an equilibrium mixture of parent–daughter radionuclide pair in an electrochemical cell under the influence of optimal applied potential. Electrodeposited daughter radionuclide is then stripped back into a 0.9 % saline solution
7.6 Key Examples of Therapeutic Radioisotopes Available from Radionuclide Generator Systems
As summarized in Table 7.1, several therapeutic radioisotopes of current interest are available from radionuclide generator systems. Although most of these systems provide beta-particle- or alpha-emitting radioisotopes, there are a very limited number of generator systems which provide Auger electron-emitting daughter radioisotopes, and only the rhodium-103 (103Rh)/ruthenium-103m (103mRu) is discussed.
7.6.1 Radionuclide Generator Systems Which Provide Beta-Emitting Radioisotopes
There a number of well-established radionuclide generator systems described in the literature which provide beta-particle-emitting daughter radioisotopes which have a broad established applications in nuclear medicine, oncology, and interventional applications
7.6.1.1 Dysprosium-166/Holmium-166 Generator
The166Dy (T ½ = 3.4 days)/166Ho (T ½ = 1.117 days) has been of interest for many years. Although 166Ho can be produced with a specific activity of only 8–9 Ci mg−1 (~300 GBq mg−1) 165Ho by the 165Ho(n,γ)166Ho reaction even at saturation in a high thermal flux of 2.5 · 1015 n cm−2 s−1, no-carrier-added 166Ho with a theoretical specific activity of about 700 Ci mg−1 (~26 TBq mg−1) is obtained by decay of 166Dy. The 166Dy is reactor-produced by the 164Dy(n,γ)165Dy(n,γ)166Dy reaction series. Separation of 166Ho from 166Dy has traditionally been a challenge. Although not a true generator system—since the 166Dy parent is also recovered and requires column reloading for subsequent 166Ho/166Dy separations following adequate 166Ho in-growth—a method has been described for successful 166Ho/166Dy separation which uses an HPLC reversed phase ion-exchange chromatographic method utilizing Dowex AG 50WX12 or Aminex A5 cation exchangers by elution with α-hydroxy-isobutyric acid (α-HIB) of 0.085 M at pH 4.3. This approach provides a Dy/Ho separation factor of approximately 103, with subsequent purification of the 166Ho3+ by cation-exchange column chromatography after acid decomposition of the Ho-α-HIB complex (Dadachova et al. 1995a, b). More recently, an apparently simple and efficient method has described separation on the Eichrom Ln resin by elution with dilute nitric acid (Ketring et al. 2002).
Although 166Ho has attractive radionuclidic properties as a lanthanide for various vector labeling and therapeutic strategies, it has not yet been widely used. Examples are studies of protein labeling with the CHX-B-DTPA ligand system (Dadachova et al. 1997). Animal studies demonstrated that no translocation of the 166Ho daughter radionuclide occurred when the [166Ho]DTPA complex was administered to rats (Smith et al. 1995). The results of these studies may suggest that the 166Dy/166Ho system may have some promise as an in vivo generator. Clinical applications which have been recently reported include radiation synovectomy with 166Ho-ferric hydroxide (Ofluoglu et al. 2002), the use of [166Ho]DTPA liquid-filled balloons for the inhibition of restenosis following coronary angioplasty (Hong et al. 2002), and the use of [166Ho]DOTMP for myeloablative therapy of multiple myeloma (Rajendran et al. 2002). If the availability of no-carrier-added 166Ho becomes a reality, use of this generator system may be a source for these and other applications.
7.6.1.2 Tungste-188/Rhenium-188 Generator
Rhenium-188 (T 1/2 = 16.9 h) is one of the useful radionuclides for therapy emitting β− particles (2.12 MeV, 71.1 % and 1.965 MeV, 25.6 %) and emits a gamma photon at 155 keV (15.1 % abundance), which can be readily imaged with gamma cameras, providing important information on biodistribution and excretion kinetics to assess targeting and dosimetry. Rhenium-188 is used for a host of therapeutic applications (Pillai et al. 2012; Jeong and Knapp 2008; Jeong and Chung 2003; Lambert and de Klerk 2006; Iznaga-Escobar 2001). The beta energies are on the higher side giving a mean range of ~3.5 mm in soft tissues. The 155 keV (14.9 %) gamma rays are suitable for imaging. The issues associated with reactor production of the 188W parent are discussed in Chap. 5.
Extensive research on 188W production and development of the 188W/188Re generator system was initiated at the Oak Ridge National Laboratory (ORNL) in the mid 1980s, focused on preparation of a 188W/188Re generator system using acidic alumina matrix analogous to the widely used 99Mo/99mTc generator (Knapp et al. 1991, 1993, 1997; Kamioki et al. 1994; Jeong and Knapp 2008). Owing to the limited sorption capacity of alumina (maximum 50 mg W/g) (Jeong and Knapp 2008; Pillai et al. 2012), 188Re available from the alumina-based 188W/188Re generators is of low radioactivity concentration, if low-specific-activity 188W is used. This in turn would require post-elution concentration of the 188Re eluate (Lisic et al. 1992; Knapp et al. 1998; Guhlke 1998; Guhlke et al. 2000; Sarkar et al. 2009; Mushtaq et al. 2007; Chakravarty et al. 2010a, b, c). The tremendous prospects associated with the use of generator-produced 188Re have led to development of automated systems for the concentration of 188Re eluate (Jäckel et al. 2005; Wunderlich et al. 2008). Over the years, different approaches such as the gel generator, electrochemical generator, thermo-chromatographic generator, and chromatographic generator with high-capacity adsorbents and nanomaterial-based adsorbents have been explored (Dash and Knapp 2015) which in turn has opened the prospect of using 188Re for therapy.
Rhenium-188 is currently of broad interest for the development of a wide variety of new therapeutic approaches for applications in nuclear medicine, oncology, and even interventional cardiology, because of the benefits, advantages, and significantly reduced expense of using 188Re in comparison to many other therapeutic radionuclides (Knapp et al. 1997, 1998a, b). There is currently a large number of physician-sponsored clinical protocols in progress. Tungsten-188 is reactor-produced by double neutron capture of enriched 186W by the 186W(n,γ)187W(n,γ)188We route. Since the production yield is a function of the square of the thermal neutron flux, even at high thermal neutron flux (>2 · 1015 n cm−2 s−1), 188W is produced with only modest specific activity of 4–5 Ci g−1 (~180 GBq g−1) per cycle (Knapp et al. 1994). This is due to the modest neutron capture cross sections and burnup of the 188W product (Mirzadeh et al. 1997). Although the first prototype 188W/188Re generators were described as early as 1966 using zirconium oxide (Lewis 1966) and with aluminum oxide in 1972 (Mikheev et al. 1972), in spite of the excellent radionuclidic properties of 188Re, there was no practical use of this therapeutic radionuclide until nearly 25 years later, when appropriate carrier molecules and targeting agents became available (Knapp et al. 1997, 1998a, b).
Related posts:
 Radionuclide Synovectomy: Treatment of Inflammation of the Synovial Joints
Radionuclide Synovectomy: Treatment of Inflammation of the Synovial Joints
 Introduction: Radiopharmaceuticals Play an Important Role in Both Diagnostic and Therapeutic Nuclear Medicine
Introduction: Radiopharmaceuticals Play an Important Role in Both Diagnostic and Therapeutic Nuclear Medicine
 Inhibition of Arterial Restenosis Following Balloon Angioplasty
Inhibition of Arterial Restenosis Following Balloon Angioplasty
 Therapeutic Radiopharmaceuticals for Treatment of Primary and Metastatic Hepatic Cancer
Therapeutic Radiopharmaceuticals for Treatment of Primary and Metastatic Hepatic Cancer
 Radioimmunotherapy (RIT)
Radioimmunotherapy (RIT)
 Reactor-Produced Therapeutic Radionuclides
Reactor-Produced Therapeutic Radionuclides
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


