Radiotracer Chemistry
Joanna S. Fowler
Yu-Shin Ding
Over the past 35 years, advances in radiotracer chemistry and positron emission tomography (PET) instrumentation have merged to make PET a powerful scientific tool for studying biochemical transformations and the movement of drugs in the human brain and other organs in the body. With advances in the sequencing of the human genome, we can anticipate the identification of many new genes and their protein products. This, in turn, will create the need for new radiotracers to characterize the functional activity of these new proteins in living systems, and ultimately in humans. In addition, new knowledge on progenitor cells and their promise in treating human disease calls for expanding radiotracer development so that imaging can be used to study cell trafficking as well as the molecular processes involved in stimulating these cells to differentiate in vivo.
Radiotracer chemistry is a subfield of chemistry underpinning the development of radiotracers labeled with the short-lived positron emitters, and there have been many chapters, monographs, and review articles on this PET radiotracer development (1,2,3,4,5,6,7,8). Of special utility is a recent compilation with references and compound structures of most of the PET labeled compounds classified according to compound type (9).
This chapter will update the chapter on chemistry in the first edition of this book (10), providing background on the design and synthesis of PET radiotracers and giving examples that illustrate general principles, rather than providing a comprehensive survey. This chapter will substantially focus on carbon-11 (11C) and fluorine-18 (18F), the two positron emitters at the heart of molecular imaging and medical application. There are several other important tracers that are emerging in their application, which are also briefly discussed. This chapter is organized by sections on different classes of labeled compounds designed to bind to and map specific molecular targets such as receptors, transporters, and enzymes. This chapter will conclude with a section on future outlook and needs.
Rapid Radiotracer Chemistry
Time dominates all aspects of a PET study, particularly the synthesis of the radiotracer. PET radiotracers must be synthesized and imaged within a time frame compatible with the half-life of the isotope (11). For 11C, this is typically about 10 minutes for isotope production (cyclotron bombardment), 40 minutes for radiotracer synthesis, and up to about 90 minutes for PET imaging. Thus the entire study from the end of cyclotron bombardment to the end of an imaging session must be orchestrated and carried out within about 2.5 hours. Large amounts of radioactivity need to be used initially in order to compensate for radioactive decay and for the sometimes low synthetic yields. Shielding, remote operations, and automation are integrated into the experimental design (12,13). It is ideal to introduce the radioactivity at the last step in the synthesis, which may require multistep syntheses of suitably protected substrates into which the 11C or 18F can be introduced. When protective groups are used, they must be stable to the labeling conditions, and the deprotection conditions must be rapid. The crude reaction mixture is usually purified by high-performance liquid chromatography or a combination of solid phase extraction and high-performance liquid chromatography. Since radiotracers are typically administered intravenously, procedures must be developed to yield radiotracers that are not only chemically and radiochemically pure but also sterile and free from pyrogens (14). One of the exciting new advances is the integration of microfluidics into radiotracer research and routine production (15,16).
Carbon-11 and Fluorine-18 Production
Basic research in hot atom chemistry provided some of the knowledge to understand the chemistry that takes place during the production of the short-lived positron emitters, and it set the stage for
producing the short-lived positron emitters in chemical forms, which were useful for the synthesis of complex radiotracers (for a review see Wolf and Redvanly (17)). Because of the short half-lives of 11C and 18F, each radiotracer synthesis requires the production of the isotope and its conversion to a useful labeled precursor molecule either directly or via some postirradiation synthesis. Production involves bombarding appropriate stable (and sometimes enriched) isotopes with charged particles such as protons and deuterons, which are most commonly and conveniently produced using a cyclotron. Three nuclear reactions, the 14N(p,α)11C, the 18O(p,n)18F, and the 20Ne(d,α)18F reactions are most commonly used for 11C and 18F production (Fig. 2.1)(2).
producing the short-lived positron emitters in chemical forms, which were useful for the synthesis of complex radiotracers (for a review see Wolf and Redvanly (17)). Because of the short half-lives of 11C and 18F, each radiotracer synthesis requires the production of the isotope and its conversion to a useful labeled precursor molecule either directly or via some postirradiation synthesis. Production involves bombarding appropriate stable (and sometimes enriched) isotopes with charged particles such as protons and deuterons, which are most commonly and conveniently produced using a cyclotron. Three nuclear reactions, the 14N(p,α)11C, the 18O(p,n)18F, and the 20Ne(d,α)18F reactions are most commonly used for 11C and 18F production (Fig. 2.1)(2).
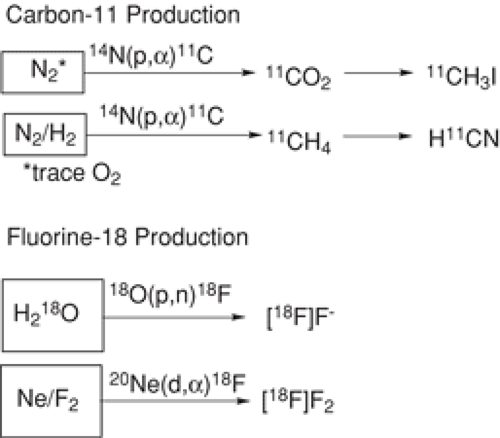 Figure 2.1. Common nuclear reactions and target materials for carbon-11 and fluorine-18 production. |
An important point is that the substrate that is used for the nuclear reaction (referred to as the target) is usually a different element than the radioisotope produced. Carbon-11, for example, is usually produced from the cyclotron bombardment of stable nitrogen (14N(p,α)11C). In principle, at the end of cyclotron bombardment, the only isotope of carbon present would be 11C. However, because stable carbon is ubiquitous in nature, it is not possible to remove it completely from the target and from the reagents used in the synthesis. Even with this unavoidable dilution, the specific activity (units of radioactivity/unit of mass) of PET radiotracers is quite high (Table 2.1). Thus it is typical that the 12C:11C ratio is about 5,000:1, which generally produces radiotracers of sufficiently high specific activity for tracer studies.
Table 2.1 Physical Properties of the Short-Lived Positron Emitters | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||
Fluorine-18 is most commonly produced by bombarding oxygen-18 enriched water with protons to yield [18F]fluoride (18). As is the case with 11C, it is not possible to remove all stable fluoride ions from the target materials and from the reagents used in the synthesis so that the isotope is always diluted with stable fluoride ion. In contrast to [18F]fluoride, which is always produced without the intentional addition of stable fluoride, [18F]F2 is always deliberately diluted with unlabeled F2. In general, for equal amounts of radioactivity, the chemical mass associated with an [18F]F2 derived radiotracer (carrier-added) exceeds that of an [18F]fluoride ion derived radiotracer (no-carrier-added) by a factor of 1,000 (3). However, there has been progress in achieving high specific activity 18F labeled elemental fluorine by mixing no carrier added (NCA) 18F labeled methyl fluoride (synthesized from aqueous [18F]fluoride ion) with a small amount of elemental fluorine in an inert neon matrix and subjecting it to an electrical discharge (19). The resulting labeled elemental fluorine has a specific activity of up to 55 GBq/μmol, which is sufficiently high for tracer studies of the dopamine transporter (20). Although [18F]XeF2 has been prepared and used in labeling, most labeling requires the use of [18F]F2 (3). Recently it has been reported that XeF2 exchanges with [18F]fluoride under mild conditions (21); however, specific activity has not been optimized.
High specific activity radiotracers provide the opportunity for imaging biological targets such as neurotransmitter receptors at tracer concentrations (22). However, the chemical mass that constitutes a tracer dose depends on the process being measured. For example, biological targets such as neurotransmitter receptors occur at much lower concentrations than enzymes, and thus higher specific activities are required for neurotransmitter or steroid hormone receptor studies. Typically specific activity is expressed by one of the following terms (23,24):
Carrier free (CF) should mean that the radionuclide or stable nuclide is not contaminated with any other radio or stable nuclide of the same element. This has probably never been achieved with 11C or 18F tracers.
No carrier added (NCA) should apply to an element or compound to which no carrier of the same element or compound has been
intentionally or otherwise added during its preparation. This applies to most radiotracers.
Carrier added (CA) should apply to any element or compound to which a known amount of carrier has been added.
Because the reporting of specific activities has ambiguities especially in the older literature, other descriptive terms have been introduced to describe radiotracer specific activities, (3) including the term effective specific activity, which refers to the value of specific activity measured by biological or biochemical assay and takes into account the presence of species that have biological effects similar to the parent compound. The factors influencing the values of specific activities of compounds that are determined physicochemically and by radioreceptor techniques have been discussed elsewhere (25).
The high specific activity of 11C and 18F labeled precursors influences the stoichiometry and the scale of the reaction. For example, in a NCA synthesis with Na[11C]N (specific activity: 2,000 Ci/mmol), the quantity of NaCN used if one starts with 100 mCi is 50 nmol. With this small quantity of Na[11C]N, all other substrates or reactants used in the synthesis are necessarily in large excess, which is problematic when an excess of a given reagent cannot be tolerated. One must consider that the substrate that will undergo reaction with the labeled precursors must be in a sufficient concentration to react both with the labeled precursors and with other competing reactants that may be present in the reaction mixture.
With such high specific activity 11C and 18F labeled precursors, syntheses are always carried out on a micro or a semimicro scale, and typical chemical masses associated with NCA PET radiotracers are a few micrograms or less. The small scale is advantageous in terms of the relative ease and speed with which one can handle small quantities of reagents and solvents, in minimizing the amounts of substances to be removed in the final purification, and in avoiding the unintentional introduction of impurities that may negatively influence the course of the reaction. However, losses caused by surface-to-volume effects, by adsorption properties of vessels, and of materials used for purification can impact heavily on yields.
Carbon-11 Labeled Compounds
The advantages of 11C as a label are many, including that it can substituted for stable carbon in an organic compound without changing the properties of the molecule. This is of particular importance for the use of PET in drug research and development (26,27). In addition, PET studies can be repeated at 2-hour intervals with a 11C labeled tracer, allowing baseline and experimental studies to be carried out in a single individual within a short time frame. However, there are large experimental hurdles imposed by the 20.4-minute half-life and the limited number of labeled precursors available for synthesis. More specifically, only [11C]O2 and [11C]H4 come directly from the cyclotron target using properly adjusted radiation conditions (Fig. 1.1). A number of other precursor molecules are synthesized from labeled carbon dioxide or methane, but all require some synthetic manipulation during or after cyclotron bombardment (2).
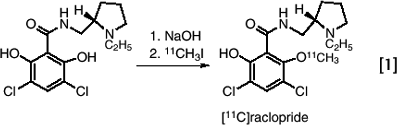
Some of the earliest syntheses with 11C depended directly on labeled carbon dioxide and hydrogen cyanide (28). Today, however, alkylation with [11C]methyl iodide is the most widely used method for introducing 11C into organic molecules (29). Alkylations are generally straightforward as in the case of the synthesis of [11C]raclopride, a widely used radiotracer for imaging dopamine D2 receptor, which is synthesized by alkylating the nor-compound with [11C]methyl iodide (30). (eq. 1)
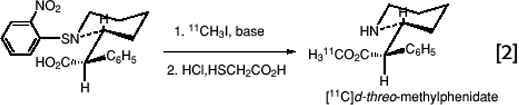
Frequently, however, reactive centers on the reaction substrate must be masked with protective groups that can be rapidly removed. This is illustrated by the synthesis of [11C]d-threo (or l- threo-methylphenidate) from labeled methyl iodide and a protected derivative of d– or l-threo ritalinic acid (31). (eq. 2)
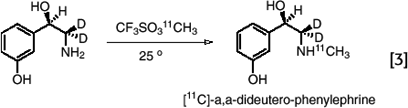
In the case of relatively sensitive compounds like deuterium- substituted [11C]phenylephrine, alkylation can be carried out under milder conditions using [11C]methyl triflate (32,33). (eq. 3)
The introduction of an online gas phase synthesis to give high specific activity [11C]H3I from labeled methane is a major advance (34,35). Other precursors from [11C]methyl iodide include [11C]methyl magnesium iodide (36) and the Wittig reagent [11C]methylenetriphenylphosphorane (37).
Many useful 11C tracers cannot be synthesized simply by either O- or N-alkylating the nor-compound with [11C]methyl iodide. For examples, the synthesis of PHNO and NPA, potential radiotracers for in vivo imaging of the dopamine D2 high-affinity state, requires the N-alkylation with 11C-labeled propionyl chloride (prepared from [11C]O2 and ethyl magnesium bromide), followed by reduction with lithium aluminum hydride (38,39). The synthesis of 11C GR89696 or its active enantiomer GR103545 (k-opioid receptor ligand) requires the N-alkylation with a 11C-labeled methylcarbonyl group, which is prepared by the reaction of 11C methanol and phosgene (40).
Carbon-11 synthesis is frequently complicated by the need for chiral-labeled products. In the case of radiotracers like [11C]d-threo-methylphenidate described above (eq. 2), this is readily accomplished because the chiral center is present in the substrate (d-ritalinic acid) and the reaction conditions preserve the chirality. Chiral high- performance liquid chromatography can also be used to separate the desired labeled enantiomer from a labeled racemic mixture. Asymmetric syntheses have been developed to directly obtain the desired
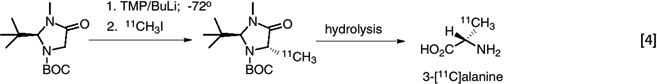
enantiomer. For example, enantiomerically enriched 3-[11C]L- alanine was synthesized from [11C]O2 (via methyl iodide) (41). (eq. 4)
enantiomer. For example, enantiomerically enriched 3-[11C]L- alanine was synthesized from [11C]O2 (via methyl iodide) (41). (eq. 4)
Labeled methane is also very useful in synthesis because it is available in large quantities and can be readily converted to H[11C]N by passing the target gas (N2/H2 plus a small quantity of ammonia) over platinum wool at 1,000°C (28). It has been used in the synthesis of labeled amines, ketones, aldehydes, acids, and amino acids (3).
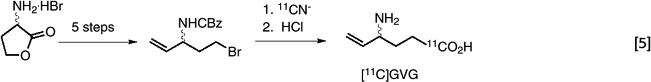
A special problem in 11C (and 18F) synthesis is the need to design synthetic routes to precursor molecules that are amendable to rapid labeling. Frequently, a new synthetic strategy must be developed for these molecules. The synthesis of [carboxyl-11C]-γ-vinyl-γ-amino-butyric acid ([11C]GVG) required the development of a new five-step synthetic strategy to prepare a suitably protected labeling precursor for the displacement reaction (42). (eq. 5)
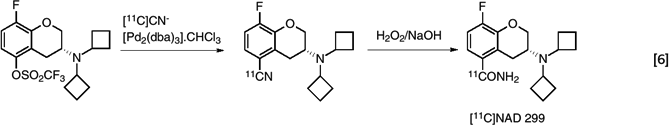
In another example, the palladium catalyzed coupling of labeled cyanide to an aromatic ring was used in the synthesis of a radioligand [11C]NAD-299 for evaluation of binding to the 5-HT1A receptor (43). (eq. 6)
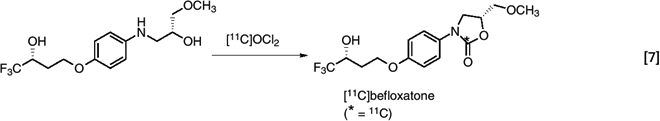
Labeled methane has also been converted to [11C]phosgene, which has been used in the synthesis of many compounds including a ring-labeled monoamine oxidase inhibitor befloxatone (44). (eq. 7)
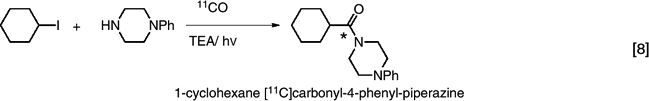
Labeled carbon monoxide ([11C]O) has also been used in the photoinitiated carbonylation reaction to produce carbonyl-11C-labeled amides (45). (eq. 8)
Problems in 11C synthesis generally include rigorously excluding stable carbon in order to maximize the specific activity of the product, optimizing reaction rates, and developing rapid methods to separate the labeled product from the starting materials and by-products. Reaction times have been reduced and yields have been increased for many labeled compounds by applying microwave technology (46).
Fluorine-18 Labeled Compounds
In contrast to the 20-minute half-life of 11C, which requires that the entire synthesis be accomplished in about 40 minutes, the half-life of 18F at 110 minutes allows more time for relatively complex synthetic manipulations and for more lengthy biological studies. An additional advantage is that 18F has a low positron energy, and thus its maximum range (2.4 mm) allows for the sharpest imaging with a high-resolution PET (Table 2.1). Moreover, as has been exemplified by the successful distribution of 2-deoxy-2-[18F]fluoro-D-glucose (18FDG) from regional production centers, 18F, when tagged to well-designed and validated radiotracers, offers the potential for distribution from a regional production center to clinical and research institutions without a cyclotron and chemistry group. Disadvantages of 18F relative to 11C include the fact that fluorine is not normally present in biological molecules or drugs and the fact that the 110-minute half-life precludes the performance of multiple studies on the same subject on the same day.
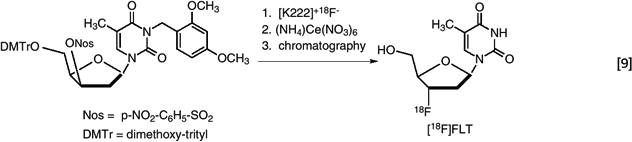
There are two simple labeled forms of 18F (fluoride ion and elemental fluorine) that are directly available for radiotracer synthesis. Fluoride ion is the more desirable of the two because it can be produced in higher yield and without added carrier. In principle, 100% of the isotope can be incorporated into the tracer. An example is synthesis of 3′-deoxy-3′-[18F]fluorothymidine ([18F]FLT) based on the [18F]fluoride displacement of a protected nosylate precursor (47). (eq. 9)
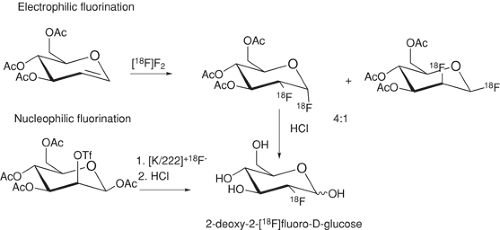 Figure 2.2. Electrophilic and nucleophilic routes to 2-deoxy-2-[18F]fluoro-D-glucose (18FDG). |
In another example, 16-α-[18F]fluoro-estradiol-17-β has been synthesized by the displacement of a triflate with [18F]fluoride and the effect [18F]fluoride solubilization conditions has been studied (48).
In contrast to [18F]fluoride displacement, the maximum radiochemical yield when [18F]F2 is used as a precursor is usually only 50%, because only one of the fluorine atoms in the fluorine molecule is labeled and typically only one atom of fluorine is incorporated into the final product after electrophilic fluorination. This loss of 50% of the label also applies when labeled fluorine is converted to other precursors like labeled acetyl hyprofluorite (3). 18FDG, which is used to measure glucose metabolism, exemplifies the evolution and improvement of a synthesis through time (Fig. 2.2) (for review see Fowler and Ido. [49]). 18FDG was first synthesized by the electrophilic fluorination with [18F]F2 and an improved synthesis from [18F]acetylhypofluorite was reported a few years later (50). However, the synthesis of 18FDG via a nucleophilic substitution with [18F]fluoride was a major breakthrough (51). It gave significantly higher yields and has largely replaced the electrophilic route. 18FDG, the most widely used PET tracer, is currently synthesized via a nucleophilic substitution, followed by acid or base hydrolysis (49). Automated 18FDG synthesis modules produce 18FDG efficiently in research labs or commercial entities and allow it to be widely distributed to other research institutes for carrying
out basic and clinical research in humans. Although some radiotracers such as [18F]fluorodopa (6-[18F]fluoro]-3,4-dihydroxyphenylalanine) (52) are still conveniently synthesized from [18F]F2 or [18F]acetylhypofluorite (53), most 18F-labeled radiotracers including [18F]fluorodopa can now be prepared from [18F]fluoride ion (54,55).
out basic and clinical research in humans. Although some radiotracers such as [18F]fluorodopa (6-[18F]fluoro]-3,4-dihydroxyphenylalanine) (52) are still conveniently synthesized from [18F]F2 or [18F]acetylhypofluorite (53), most 18F-labeled radiotracers including [18F]fluorodopa can now be prepared from [18F]fluoride ion (54,55).
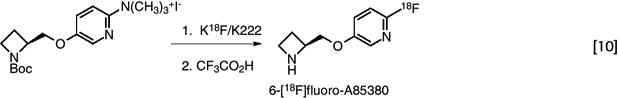
The most successful approach for preparing high specific activity 18F-substituted aromatic compounds is the nucleophilic aromatic substitution reaction (56). The minimal structural requirements for efficient nucleophilic aromatic substitution are the presence of an electron withdrawing, activating substituent such as RCO, CN, NO2 and so forth, as well as a leaving group, such as nitro- or trimethylammonium. This approach is used for the high-yield one pot, two-step synthesis of 6-[18F]fluoro-A85380, a radiotracer with high specificity for nicotinic acetylcholine receptors (57,58,59). (eq. 10)
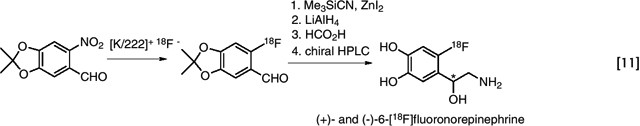
In addition to simple fluorine-substituted aromatic compounds, there are important radiotracers with electron donating substituents on the aromatic ring that can impede the nucleophilic aromatic substitution reaction. Recent mechanistic studies have established that nucleophilic aromatic substitution can be carried out in the presence of suitably protected electron donating groups, thus extending the utility of the nucleophilic aromatic substitution to the synthesis of 6-[18F]fluorodopa as well as 6-[18F]fluorodopamine (60), (+)- and (–)-6-[18F]fluoronorepinephrine (61). (eq. 11)
The use of the nucleophilic aromatic substitution reaction produced 6-[18F]fluorodopamine in sufficiently high specific activity for tracer studies without producing hemodynamic effects (62), which are observed when low specific activity 6-[18F]fluorodopamine is used.
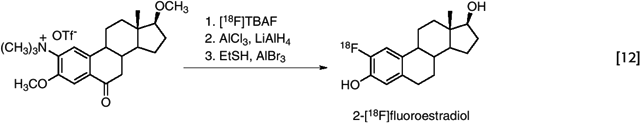
In another example of nucleophilic aromatic substitution on an electron-rich ring, 2-[18F]fluoroestradiol was synthesized by the displacement of a trimethylammonium group on the carbon-2 (2C) of an estrogen derivative, with additional activation being provided by a 6-keto group, which was subsequently removed by reduction (63). (eq. 12)
Simple, no-carrier-added [18F]fluoroarenes have also been synthesized via the reaction of [18F]fluoride with diaryliodonium salts (64).
Radiotracer Design and Mechanisms
Radiotracer design refers to the process of choosing priority structures for labeling in order to image a specific molecular target. For this purpose, it is essential to consider the affinity and selectivity of the compound for the molecular target as well as the concentration of the molecular target itself (for a discussion see Eckelman et al. [65]). However, a chemical compound with high affinity and high selectivity for a specific molecular target that is present in high concentration will not necessarily guarantee high binding specificity and appropriate kinetics in vivo. This is due to the fact that the human and animal body presents a complex set of barriers that compete with specific binding and with radiotracer delivery to the target organ. These include plasma protein binding (for examples see Yu et al. [66] and Ding et al. [67]) sequestration in cells, low affinity and nonspecific binding, metabolism, the blood–brain barrier, and probably many others.
For most radiotracers, the physicochemical properties of the compounds, such as their size, charge, solubility, and lipophilicity, are useful parameters and are particularly important in the design of tracers that penetrate the blood–brain barrier (68,69,70). Thus, the literature of medicinal and pharmaceutical chemistry is a valuable resource for the design and evaluation process of radiotracers (71).
The design of 18FDG is a good example of radiotracer design that benefited a great deal from information that was in the literature at the time. 18FDG was modeled after carbon-14 labeled 2-deoxy-glucose (14C-2DG), a tracer used to measure brain glucose metabolism in animals using autoradiography (Fig. 2.3) (72). The requirements for a positron emitter labeled version of 2-deoxyglucose were:
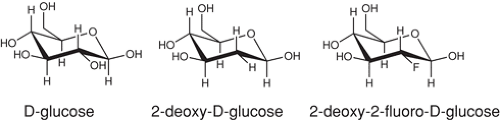 Figure 2.3. Structures of glucose, 2-deoxy-D-glucose and 2-deoxy-2-fluoro-D-glucose. |
The resulting molecule must contain a positron emitter.
The resulting molecule must be a substrate for the glucose transporter, which facilitates the transport of glucose from blood to brain.
The resulting molecule must be a substrate for hexokinase the pivotal enzyme in glycolysis.
The resulting molecule must not undergo metabolism past the point of phosphorylation, allowing the phosphorylated product to be trapped at the site of phosphorylation.
As was reported in 1954, the hydroxyl group on 2C-2 on the glucose skeleton is the only hydroxyl group on glucose that can be removed and still retain the ability of the molecule to be a substrate for hexokinase (typically the rate-limiting enzyme in glycolysis) (73). For this reason, 2C-2 was selected for fluorine substitution. Moreover, the glucose transporter is not sensitive to substitution on 2C-2, and the metabolism beyond the hexokinase step requires the hydroxyl group in 2C-2. Thus 18FDG, in which the 18F is on 2C-2, undergoes carrier mediate transport into the brain where it is phosphorylated to 18FDG-6-phosphate by hexokinase. However, because the hydroxyl group in 2C-2 is missing, no further metabolism occurs and 18FDG-6-phosphate is trapped in the cell where metabolism has occurred (Fig. 2.4). When imaged, metabolically trapped 18FDG-6-phosphate provides a map of glucose metabolism in all brain regions simultaneously. [1-11C]2-deoxy-D-glucose was also labeled with 11C shortly after the development of 18FDG and provided an opportunity to do multiple studies on the same subject in a single scanning session (74,75).
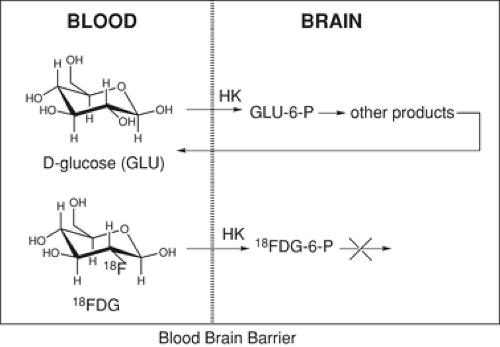 Figure 2.4. Simplified diagram comparing the behavior of glucose and 2-deoxy-2-[18F]fluoro-D-glucose (18FDG) in the brain. Glucose crosses the blood–brain barrier by facilitated transport and enters a cell. There it enters the glycolytic cycle where it is first phosphorylated by hexokinase (HK) and eventually produces adenosine triphosphate and metabolites that can leave the cell. Like glucose, 18FDG also undergoes facilitated transport into the brain and is phosphorylated by hexokinase to produce 18FDG -6-phosphate (18FDG-6-P). However 18FDG-6-P does not undergo further metabolism and is trapped in the cell. |
Although 18FDG was initially developed for imaging brain metabolism, early studies showed that it was concentrated in animal tumors (76) due to enhanced glycolysis in cancer cells (77). Consequently, 18FDG has had a profound impact on the management of patients with cancer (78). The success of 18FDG in the detection of human cancers also relates in part to the replacement of the hydroxyl on 2C-2 rather than on another carbon atom. Because the hydroxyl group on 2C-2 is required for active transport of glucose and because the active transport of glucose across the renal tubule is required for glucose resorption (79), 18FDG, in contrast to glucose, is excreted. This, in effect, it lowers the body background after the injection of 18FDG, creating the high contrast for imaging metabolically active tumors.
Radiotracer Validation
A key step in the development of a new radiotracer is to characterize its binding in vitro and more important, in vivo. Since the PET image itself originates from the annihilation photons produced by positron decay, it provides no information about the chemical compound(s), giving rise to the image or on the cellular or subcellular localization or binding site or on binding specificity. One way to ensure specificity and saturability is to compare the distribution and kinetics of the radiotracer before and after treatment with a pharmacologically specific drug or with a pharmacological dose of the tracer molecule. Another tool for assessing specificity is to compare the binding of the labeled compound in wild type and knockout animals in which the gene for a specific molecular target (receptor, transporter, enzyme) is removed (80). Other approaches include examining the behavior of the same labeled compound in different positions, the comparison of labeled stereoisomers, and, in limited cases, the use of deuterium isotope effects to probe specific reactions (81). Radiotracer kinetics can also be a limiting factor in quantification, therefore, they must be critically examined and appropriate kinetic models developed to calculate parameters that can be related to receptor concentration, enzyme activity, or some other factor (82,83,84,85,86,87,88). Some examples are given for illustration.
Comparative Studies of the Same Molecule Labeled in Different Positions
Frequently the position of the label can be adjusted to simplify the profile of labeled metabolites contributing to the image or to determine
the localization mechanism. For example, high pancreas uptake of 11C was observed after the injection of [11C]L-dopa (89). To determine the mechanism by which 11C concentrates in the pancreas, a comparison of the radiotracer labeled in the carboxyl group and in the β position was made in the same subject. Only the radiotracer labeled in the β position was retained in the pancreas. This demonstrated that aromatic amino acid decarboxylase (AAADC) (which would result in the loss of the 11C in the carboxyl position, but not the β position) was responsible for the retention of [11C]L-dopa in pancreas and pancreatic tumors. A similar comparative study of [11C]5-hydroxytryptophan (5-HP) labeled in the β position and in the carboxyl position demonstrated tumor uptake and decarboxylation and also provided the means for quantitating AAADC activity. A PET study of [11C]dimethylphenethylamine labeled in two different positions (N-methyl vs. α-methylene) also illustrates the profound difference in 11C kinetics depending on the position of the label, illustrating the importance of label position for probing enzyme activity (90). [11C]WAY-100,635 which has been developed as a radiotracer for the 5-HT1A receptor, was initially labeled in the methoxy position but was subsequently labeled in the carbonyl position to reduce the contribution of labeled metabolites (91). Structures of some of these labeled compounds are shown in Fig. 2.5.
the localization mechanism. For example, high pancreas uptake of 11C was observed after the injection of [11C]L-dopa (89). To determine the mechanism by which 11C concentrates in the pancreas, a comparison of the radiotracer labeled in the carboxyl group and in the β position was made in the same subject. Only the radiotracer labeled in the β position was retained in the pancreas. This demonstrated that aromatic amino acid decarboxylase (AAADC) (which would result in the loss of the 11C in the carboxyl position, but not the β position) was responsible for the retention of [11C]L-dopa in pancreas and pancreatic tumors. A similar comparative study of [11C]5-hydroxytryptophan (5-HP) labeled in the β position and in the carboxyl position demonstrated tumor uptake and decarboxylation and also provided the means for quantitating AAADC activity. A PET study of [11C]dimethylphenethylamine labeled in two different positions (N-methyl vs. α-methylene) also illustrates the profound difference in 11C kinetics depending on the position of the label, illustrating the importance of label position for probing enzyme activity (90). [11C]WAY-100,635 which has been developed as a radiotracer for the 5-HT1A receptor, was initially labeled in the methoxy position but was subsequently labeled in the carbonyl position to reduce the contribution of labeled metabolites (91). Structures of some of these labeled compounds are shown in Fig. 2.5.
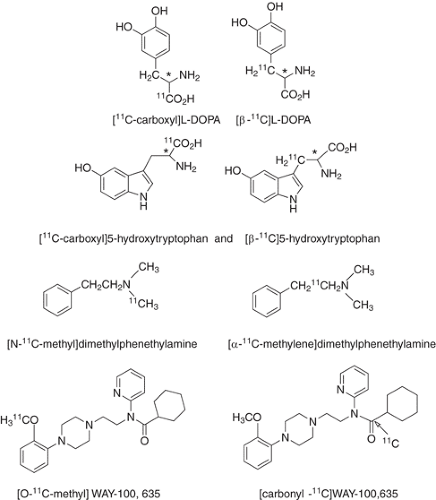 Figure 2.5. Structures of labeled compounds in which different label positions have been synthesized and compared either to obtain mechanistic information or to simplify the interpretation of the PET image. |
Comparison of Labeled Stereoisomers
Biological targets (enzymes, receptors, etc.) are chiral molecules, and frequently chiral ligands and drugs show binding selectivity for one enantiomer over the other. In some cases, this is useful in assessing binding specificity. For example, methylphenidate (Ritalin) is a racemic drug that binds to the dopamine transporter, and it is used in the treatment of attention deficit hyperactivity disorder and narcolepsy. Comparative PET studies of two enantiomers, [11C]d-threo-methylphenidate and [11C]l-threo-methylphenidate, provided confirmatory evidence that the pharmacological activity of the drug resides in the d-threo-enantiomer, which binds to the human striatum, in contrast to [11C]l-threo-methylphenidate, which shows no specific retention (92). [11C]d-threo-methylphenidate proved to be an excellent radiotracer for measuring dopamine transporter availability in the human brain (93). A comparison of labeled enantiomers has also been applied in the study of radiotracers for the muscarinic cholinergic system, [11C]dexetimide and [11C]levetimide (94). Another example applies to the development of radiotracers for studies of the serotonin transporter. In one case comparative studies of [11C](+)McN5652 and [11C](–)McN5662 in both mice and humans showed the expected retention of the (+)-enantiomer and clearance of the (-)-enantiomer (95).
Although the comparison of active and inactive enantiomers in vivo is potentially a powerful mechanistic tool, there are pitfalls. This was encountered in the comparison of the active and inactive enantiomers of cocaine (- and + cocaine) to assess cocaine’s binding specificity to the dopamine transporter. Instead of demonstrating stereoselective binding in the brain, as was anticipated, the comparison of 11C labeled negative and positive cocaine revealed an extraordinarily rapid metabolism of [11C](+)-cocaine by butyrylcholinesterase, which prevented its penetration into the brain (96). Thus, in this case, rapid peripheral metabolism, rather than stereospecific binding to the dopamine transporter, was responsible for the observed behavior in vivo.
Deuterium Isotope Effects
A carbon–hydrogen (C-H) bond is more easily cleaved than a carbon–deuterium bond. If a specific C-H bond in a radiotracer is cleaved in the rate limiting or rate contributing step of a chemical or biochemical reaction, its substitution with deuterium will reduce the rate of reaction and the rate of accumulation in tissue. Deuterium isotope effects have been used as a mechanistic tool in radiotracer studies of monoamine oxidase (MAO), an enzyme known to exhibit strong isotope effects due to the cleavage of the C-H bond α to the nitrogen atom where oxidation is occurring (97). There are several examples in the radiotracer literature, including [11C]L-deprenyl and deuterium-substituted [11C]L-deprenyl (98); [11C]clorgyline and deuterium substituted [11C]clorgyline (99); [13N]phenethylamine and deuterium substituted [13N]phenethylamine (100); [11C]phenylephrine and deuterium substituted phenylephrine (101); and 6-[18F]fluorodopamine and deuterium substituted 6-[18F]fluorodopamine (102). The binding of these radiotracers in tissue (and the PET images) show deuterium isotope effects consistent with MAO catalyzed cleavage of a specific bond. In the case of 6-[18F]fluorodopamine, a comparison of deuterium substitution in the α and in the β positions was used to determine that MAO and not dopamine-β-hydroxylase was responsible for the kinetic profile of 6-[18F]fluorodopamine in the heart (102). In the case of [11C]L-deprenyl, the slower rate of trapping of the deuterium substituted molecule in brain facilitated kinetic modeling in humans (103). Although the deuterium isotope effect is dramatic in these cases, it is important to note that this only is useful in mechanistic studies in special cases such as MAO where C-H bond cleavage occurs in the rate-limiting step. Radiotracer structures are shown in Fig. 2.6.
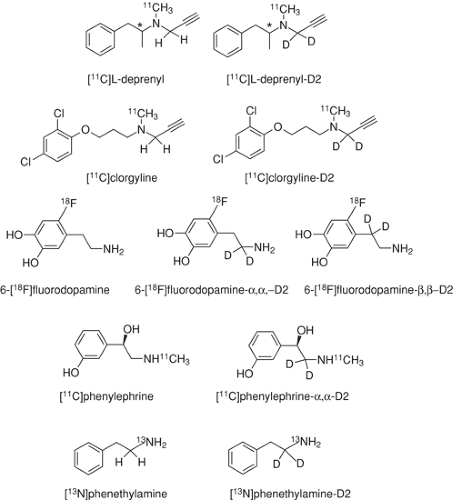 Figure 2.6. Structures of labeled compounds substituted with deuterium to obtain mechanistic information and/or to improve radiotracer kinetics. |
Radiotracers for Neurotransmitter Systems
The development of radiotracers for imaging neurotransmitter systems (including their receptors, plasma membrane, vesicular transporters, the enzymes that synthesize and degrade them, and the processes involved in signal transduction) has dominated radiotracer research over the past several decades (104). The reasons for this are obvious when one considers that brain function is driven by the complex interplay of neurotransmitter systems and that their disruption underlies many diseases of the central nervous system (105). Indeed PET has made it possible to visualize many different aspects of neurotransmitter activity in living systems. Radiotracers for some of the major neurotransmitters will be highlighted in the following sections.
The Brain Dopamine System
Dopamine plays a pivotal role in the regulation of movement, motivation, and cognition. It is also closely linked to reward and reinforcement addiction and is a major molecular target of stimulant drugs and drugs to treat Parkinson disease and schizophrenia. Drugs abused by humans also elevate brain dopamine, although by a variety of different mechanisms. Because of its importance, the study of the brain dopamine system has been a major focus in the basic and clinical neurosciences, and it is by far the most widely studied neurotransmitter system in PET research (106,107).
Dopamine is synthesized in the dopaminergic neurons in the substantia nigra, the ventral tegmental area, and the retrorubral area of the mesencephalon. It is stored within vesicles to protect it from oxidation by MAO. It is released into the synapse in response to an action potential and interacts with dopamine receptors that are present on both postsynaptic sites (where they function in cell-to-cell communication) and on presynaptic sites (where they regulate exocytotic release and dopamine synthesis). There are five subtypes of dopamine receptors that are grouped in two major families: those that stimulate adenyl cyclase (D1, D5) and those that inhibit it (D2, D3, D4) (108). The dopamine D1 and D2 receptors have significantly higher concentrations than the other subtypes and have highest density in the striatum. Quantitative PET studies of the brain dopamine system are facilitated by the high concentration of dopamine receptors and dopamine transporters in the basal ganglia and an absence of these elements in the cerebellum.
The synaptic concentration of dopamine is regulated primarily by reuptake. Dopamine release is regulated by presynaptic autoreceptors as well as by other neuroanatomically distinct neurotransmitters through interactions with the dopamine neuron. Dopamine is also removed by oxidation by MAO and methylation by catechol-O-methyltransferase (COMT).
Dopamine Metabolism
Dopamine does not cross the blood–brain barrier, and thus the investigation of brain dopamine metabolism with PET required a labeled derivative of L-dopa, the amino acid precursor of dopamine, which is transported into the brain via the large neutral amino acid carrier. [18F]Fluorodopa was the first radiotracer developed to probe brain dopamine metabolism (109). 6-[18F]fluorodopa crosses the blood–brain barrier and is accumulated in dopamine terminals and converted to 6-[18F]fluorodopamine (110). Decreased [18F]fluorodopa accumulation is associated with the loss of dopaminergic neurons (111). Because the metabolism of [18F]fluorodopa is complex, 18F-labeled m-tyrosine, which has simpler metabolism, has also been developed to improve quantification (112). Nonetheless, [18F]fluorodopa is the more widely used of the two. [11C]L-dopa has also been synthesized with the label in two positions and represents a labeled version of the native amino acid (Fig. 2.5) (113).
Dopamine Receptors
Although there are five dopamine receptor subtypes, only radiotracers with specificity for dopamine D2 and D1 receptors have been developed, although promising candidates for the D3 receptor subtype have recently been identified (see Fig. 2.7 for structures of some of these compounds). Dopamine D1 and D2 receptors are highly concentrated in the striatum with lower concentrations occurring in extrastriatal regions. High-affinity radiotracers are required for imaging extrastriatal receptors because of their low density. Although most of the radiotracer development work has focused on labeled dopamine receptor antagonists, agonists are also of interest because of the potential to image the high-affinity state of the receptor.
Dopamine D2 receptor availability has been examined with PET using 11C and 18F labeled antagonists to the D2 receptor. For example, N-methylspiroperidol has been labeled with 11C and with 18F and used to measure dopamine receptor availability and absolute concentration in the normal and diseased brain and to probe dopamine receptor occupancy by antipsychotic drugs (114,115,116,117). 3-(2′-[18F]fluoroethyl)spiperone and N-(3-[18F]fluoropropyl)spiperone have also been used for the quantitative estimation of D2 receptor sites (118,119), and the ethyl derivative has also been used as a reporter in gene-expression studies (120). The irreversible or near-irreversible binding of the spiroperidol derivatives has been a limitation in terms of quantitation because the receptor-binding parameter may be influenced by radiotracer delivery.
[11C]raclopride is the most widely used PET radiotracer for studies of dopamine D2 receptor availability (121,122) and for measuring changes in extracellular dopamine brought about by drugs and other stimulants. Drug-induced changes in synaptic dopamine have also been measured with [11C]raclopride, since its moderate affinity for the dopamine D2 receptor and reversible binding allow it to compete with dopamine in the synapse (123,124). Repeated measures with [11C]raclopride with an intervening challenge with dopamine-enhancing drugs like methylphenidate and amphetamine have been successfully used to assess dopaminergic function (125,126,127). High-affinity reversibly binding benzamide radiotracers such as [18F]fallypride (128,129) and [11C]FLB 457 have also been developed in order to have a longer scanning period and higher striatum:cerebellum ratios and to image extrastriatal dopamine D2 receptors that are present in low concentration.
The development of radiotracers to probe the high-affinity state of the dopamine D2/D3 receptor has been approached by a number of investigators (130,131,132). In particular, recent PET studies with (–)-N-(11C)propyl-norapomorphine show that it is a promising tracer for high affinity states of the D2/D3 receptor (133,134). Another D2/D3 receptor agonist, [11C]-(+)-PHNO (4-propyl-9-hydroxynaphthoxazine) has also been synthesized and shown to be more sensitive to changes in dopamine than [11C]raclopride (38,39).
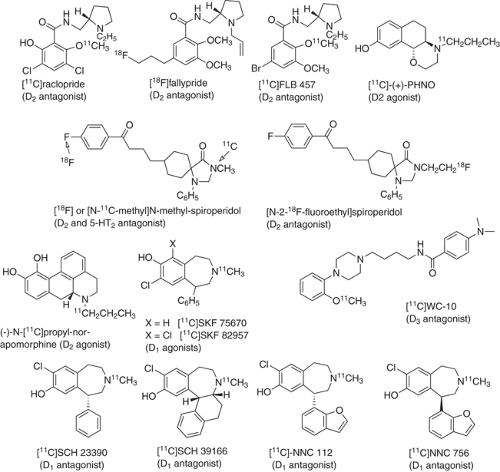 Figure 2.7. Structures of some of the radioligands developed for studies of dopamine receptors. |
The D1 receptor antagonists, SCH23390 and SCH39166, have also been labeled with 11C and used to study the central nervous system D1 receptor (135,136). A high-affinity ligand 11C labeled NNC112 has been developed for imaging extrastriatal D1 receptors in humans (137). The (+) enantiomer of NNC112 binds selectively and reversibly while the (–) enantiomer did not show appreciable binding. Studies with [11C]NNC112 in humans have shown that kinetic analysis of uptake provides an appropriate method with which to derive dopamine D1 receptor parameters both in regions of high- and low-receptor density (138).
The dopamine D3 receptor system has been implicated in a number of central nervous system disorders. It is also a new target in drug research and development (139). There has been recent effort and progress in radiotracer development (140). This has been a challenge due to the high concentrations of D2 receptors relative to D3 receptors among other factors. Recently the N-(2-methoxyphenyl)piperazine derivative WC-10 has been labeled with 11C and shown to have regional distribution consistent with binding to D3 receptors in the central nervous system (141,142).
An attempt to visualize the D4 receptor with [11C]N-[2-[4-(4-chlorophenyl)piperazin-1-yl]ethyl]-3-methoxybenzamide ([11C] PB12) was unsuccessful probably due to the low concentration of dopamine D4 receptors, insufficient binding affinity of the ligand, and high background nonspecific binding (143). Other candidate ligands have also been evaluated (144).
Dopamine and Vesicular Transporters
The plasma membrane dopamine transporter (DAT) has been studied using [11C]nomifensine (145), [11C]cocaine (146), [11C]d-threo-methylphenidate (147), cocaine analogues (148,149,150,151,152,153) and [18F]GBR13119 (154) (Fig. 2.8). These tracers and others have been applied in studies of neurodegeneration, aging, and dopamine transporter occupancy by drugs including cocaine and methylphenidate (155,156,157,158,159). [11C]Cocaine is worthy of mention. Although it was initially developed to measure cocaine pharmacokinetics in the human brain, its rapid reversible kinetics have also served to measure dopamine transporter availability using a graphical analysis for reversible systems (87). A new ligand, [11C]PE2I,
with high affinity toward dopamine transporter has been developed (160). Studies in human subjects showed radioactivity ratios of 10 and 1.8 for the striatum and substantia nigra to the cerebellum, respectively, suggesting an advantageous approach for PET examination of DAT binding in the midbrain, a region from which dopaminergic innervation originates (161). Unfortunately, the presence of the pharmacologically active metabolite confounded its kinetic analysis (162).
with high affinity toward dopamine transporter has been developed (160). Studies in human subjects showed radioactivity ratios of 10 and 1.8 for the striatum and substantia nigra to the cerebellum, respectively, suggesting an advantageous approach for PET examination of DAT binding in the midbrain, a region from which dopaminergic innervation originates (161). Unfortunately, the presence of the pharmacologically active metabolite confounded its kinetic analysis (162).
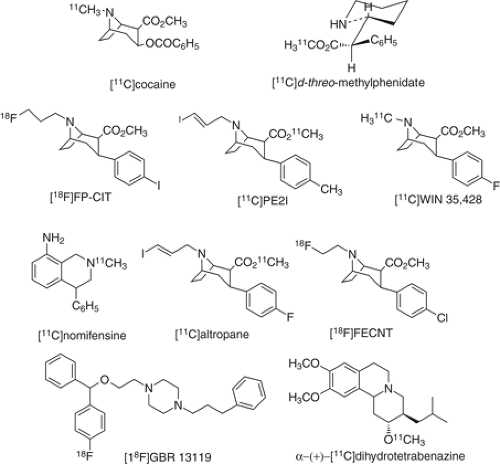 Figure 2.8. Structures of some of the labeled compounds developed for imaging the dopamine transporter and the vesicular monoamine transporter. |
Vesicular (mono)amine transporters (VMAT2) remove monoamines like dopamine from the cytosol into intracellular vesicles where they are protected from intracellular enzymes like MAO and stored for eventual exocytotic release. Studies have shown that VMAT2 is not altered by regulatory responses to nigrostriatal lesions, changes in dopamine synthesis, or turnover (163,164). Thus, VMAT2 is a useful molecular target for dopamine terminal density. Accordingly, α(+)-[11C]dihydrotetrabenazine ([11C]DTBZ) has emerged as an excellent radiotracer, devoid of lipophilic metabolites, and shows stereoselectivity and kinetics amenable to modeling (165,166). Recently, it has been shown that VMAT2 are expressed in human islet β cells as well as in tissues of the central nervous system. Diabetes results from an absolute or relative reduction in pancreatic β cell mass, leading to insufficient insulin secretion and hyperglycemia. [11C]DTBZ can be used to measure VMAT2 density and consequently provide a noninvasive measurement of β cell mass that could be used to study the pathogenesis of diabetes and to monitor therapeutic interventions (167).
Monoamine Oxidase
Monoamine oxidase (for review see Fowler et al. [168]) is a flavin-containing enzyme that exists in two subtypes: MAO A and B (see Fig. 2.9 for structures). MAO oxidizes amines including dopamine and other neurotransmitters (169). Medical interest in MAO stems from the utility of MAO inhibitor drugs in the treatment of depression and Parkinson disease. The first images of functional MAO activity in the human brain were made with 11C labeled suicide enzyme inactivators [11C]clorgyline and [11C]L-deprenyl, which covalently label the enzyme in vivo (170). Mechanistic PET studies with deuterium substituted [11C]L-deprenyl demonstrated that the C-H bond in the propargyl group was involved in the rate limiting step for the formation of the PET image and provided a means of selectively controlling the rate of trapping of tracer in the brain to improve quantitation (103). [11C]L-deprenyl and the deuterium substituted derivative have been used to directly examine the effects of MAO B inhibitor drugs, lazabemide and L-deprenyl in the human brain (171,172).
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


