and Ashutosh Dash2
(1)
Nuclear Security and Isotope Division, Oak Ridge National Laboratory, OAK RIDGE, USA
(2)
Isotope Production and Applications Division, Bhabha Atomic Research Centre, Mumbai, India
5.1 Introduction
Since most therapeutic radioisotopes used in nuclear medicine, oncology, and interventional therapeutic methods are neutron rich and often decay by beta-particle emission, they are thus generally produced in research reactors (Manual, IAEA 2003) by various reactions as shown in Fig. 5.1.
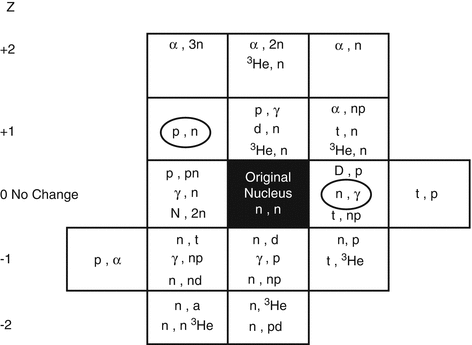
Fig. 5.1
Examples of particle-based pathways for radioisotope production
5.2 Reactor Production of Radionuclides
The neutron flux associated with nuclear fission in a reactor can be used to activate stable nuclides for radionuclide production. The target material is generally sealed in specially designed irradiation capsules which are usually pneumatically or hydraulically placed within the reactor core. These targets do not interfere with uranium fission reactions (Theobald 2011); during irradiation, the fission neutrons interact with the target material to form compound nuclei which decay via various paths producing the radioactive products.
5.3 Calculation of Production Yield
When a target is irradiated in a reactor, a nuclear reaction occurs, leading to production of isotope. The activation per second is represented by
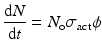
(5.1)
where N o → total no. of atoms present in target
σ act → activation cross section (cm2)
ϕ → neutron flux (neutrons.cm−2.sec−1)
This equation considers the neutron flux to be isotropic, and in the case where neutrons are not monoenergetic and a velocity distribution exists, then the average neutron flux value is considered and the neutron cross-section values are modified as required. Since the product radioisotope decays with a specific physical own half-life value, the net growth rate of radioactive product atoms can be expressed as
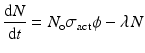
where, λ → Decay constant (sec−1) of the isotope produced
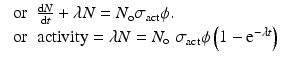
If A is the mass number of the target element under irradiation, then expression of the above equation in Curies then becomes
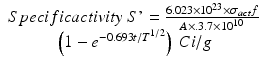
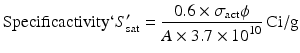
(5.2)
(5.3)
(5.4)
where σ act → activation ‘cross section (barn × 10−24)
ϕ → neutron flux (neutrons-cm−2-sec−1)
T ½ → half-life ‘of the product nucleus (sec)
t → time of irradiation (sec)
When t ≫ T ½, the production tends to saturate.
(5.5)
The above equation clearly shows that growth of activity in a target under irradiation is exponential in nature and reaches a saturation value limited by the reactor neutron flux. In practice, the activity induced in the target under irradiation will be less than the activity calculated using the above equation, due to several factors which include the following:
Target self-shielding effect in the target
Reactor power variation
Flux depression due to adjacent samples in the reactor
Target burnup over time
Destruction of the product nucleus due to subsequent neutron capture
When correction for target burnup and destruction of target atoms is considered, Eq. (5.2) becomes
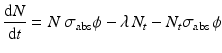
where σ abs → absorption cross section (cm2) of the target atom causing burnup,
(5.6)
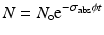
N t is the number of product atoms at time t,
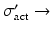 activation cross section in barn
activation cross section in barn
The above equation can then be expressed as
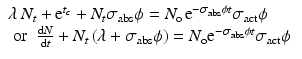
(5.7)
If 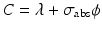 and
and 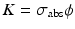 , Eq. (5.7) becomes
, Eq. (5.7) becomes
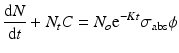
By integration and application of suitable conditions, this equation becomes
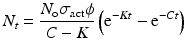
By further substituting of the factor, 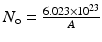 where A → atomic weight of the target atom and λN expressing the activity produced in the target, the specific activity (S) of the isotope produced will be
where A → atomic weight of the target atom and λN expressing the activity produced in the target, the specific activity (S) of the isotope produced will be
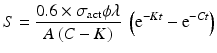
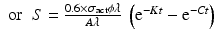
From this equation the value of time t at which the specific activity will be maximum which could be obtained can be established by equating dS/dt = 0
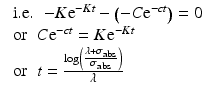
Since the target material is cooled after irradiation to allow the decay of the activated container material—which primarily consists of aluminum, i.e., 28Al and other ultra-short-lived isotopes produced—a correction factor for the cooling period (t c ) is thus introduced:
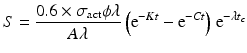
The factors which determine radionuclide production parameters in a reactor include:
and , Eq. (5.7) becomes(5.8)
(5.9)
where A → atomic weight of the target atom and λN expressing the activity produced in the target, the specific activity (S) of the isotope produced will be(5.10)
(5.11)
(5.12)
(5.13)
The energy of the neutrons and the neutron flux
The characteristics and quantity of the target material
The activation cross section for the desired nuclear reaction
Radionuclides are produced by a variety of mechanisms described below, based on different incident neutron–particle reactions as summarized in Fig. 5.1 with the target nucleus.
5.4 Direct (n, γ) Activation (Radiative Route)
The most common nuclear reaction used for reactor production of radionuclides consists of the radiative capture of thermal or epithermal neutrons by target nuclei followed by emission of prompt γ rays (Mausner et al. 1998). The activity levels produced at half saturation are related to Eq. (5.3), and by this route, the target is not entirely transformed, which results in the presence of considerable residual target atoms, or carrier, in the desired radioactive product. However, at higher neutron flux values, higher-specific-activity final products can often be produced, containing lower levels of target atoms. Thus, specific activity is a function of neutron flux and higher-specific-activity products can thus be obtained for high (n, γ) cross-section values and the target is irradiated at high neutron flux. An inherent potential disadvantage of this method is that impurities in the target will also undergo neutron activation, resulting in transformation into both radioactive and stable isotope impurities present in the desired product. For this reason, the use of target materials with very high chemical purity target is generally an essential requirement. In addition to high chemical purity, isotopically enriched targets are often also necessary in order to increase the specific activity as well as radionuclidic purity of the produced radionuclide. While production of no-carrier-added (NCA) radionuclides is not possible using the (n, γ) reaction, high-specific-activity products can be attained if the neutron capture cross sections are very high. Table 5.1 summarizes the characteristics of a variety of therapeutic radionuclides of current interest which are produced by the (n, γ) activation route.
Table 5.1
Key examples of therapeutic radionuclides produced by the (n, γ) radiative route
Target | Product | Half-life (days) |
|---|---|---|
Erbium-168 | Erbium-169 | 9.4 |
Gold-197 | Gold-198 | 2.7 |
Holmium-165 | Holmium-166 | 1.1 |
Lutetium-176 | Lutetium-177 | 6.68 |
Palladium-108 | Palladium-109 | 0.57 |
Phosphorous-31 | Phosphorous-32 | 14.3 |
Rhenium-185 | Rhenium-186 | 3.72 |
Rhenium-187 | Rhenium-188 | 0.7 |
Samarium-152 | Samarium-153 | 2.0 |
Tin-116 | Tin-117 m | 13.6 |
5.5 Neutron Activation Followed by β− Decay (n, γ → β−)
In this production mode, the target initially undergoes the (n, γ) reaction to produce an intermediate short-lived radioisotope, which on decay by beta emission produces the radionuclide of interest. The (n, γ) reaction does not directly populate the final product but represents a precursor that decays by beta decay which leads to the final product. In this case, the desired product can generally be separated from the initial product and is thus obtained with very high SA, often carrier-free. The optimum irradiation period required to attain maximum production yield depends on the half-life of the parent radionuclide. In this case, since the product radionuclide is chemically different from the original target (i.e., Z changes), the availability of efficient chemical processing is a prerequisite for obtaining the desired radionuclide. Radionuclides produced by this route are particularly attractive as they are generally NCA and will have SA values approaching the theoretical specific activity value. Typical examples of a few therapeutic radionuclides that can be produced in reactor by the (n, γ) → β− route are shown in Table 5.2. In some cases, the β−-decay product resulting from decay of the initial radioactive product can then subsequently capture a neutron to form the desired radioactive product, such as for production of 166Dy from neutron irradiation of enriched 164Dy.
Table 5.2
Examples of key therapeutic radionuclide produced by the (n, γ) → β− route
Target | Intermediate radionuclide | Product radionuclide | Half-life (days) |
|---|---|---|---|
Dysprosium-164 | Dysprosium-166 164Dy(n,γ)165Dy(n,γ)166Dy | Holmium-166 | 1.1 |
Gemanium-76 | Gemanium-77 | Arsenic-77 | 1.6 |
Neodymium-148 | Neodymium-149 | Promehtium-149 | 2.21 |
Palladium-110 | Palladium-111 m | Silver-111 | 7.45 |
Platinum-198 | Platinum-199 | Gold-199 | 3.14 |
Ruthenium-104 | Ruthenium-105 | Rhodium-105 | 1.47 |
Tellurium-130 | Tellurium-131 | Iodine-131 | 8.02 |
Ytterbium-176 | Ytterbium-177 | Lutetium-177 | 6.7 |
Xenon-124 | Xenon-25 | Iodine-125 | 60 |
5.6 The (n,p) Production Reaction
In some cases the absorption of neutron leads to emission of a proton as outgoing particle (Fig. 5.1), referred to as the (n,p) reaction. With the lower-energy thermal neutrons, the cross-section values for this reaction are quite low. Such reactions are more probable with fast neutrons having energy values above the threshold energy which is mostly between 2 and 6 MeV. While radionuclides produced by (n,p) reactions are available in NCA form, this route of production suffers from low yields since the fast flux component of neutrons in a reactor is only a fraction of the total neutron flux and fast flux component is low in many research reactors. Also in this case, the product nuclei require chemical separation from the large macroscopic levels of target atoms. Table 5.3 lists several important therapeutic radioisotopes produced by the (n,p) reaction.
Table 5.3
Examples of therapeutic radionuclides produced by the (n,p) reaction
Target nuclide | Product radionuclide | Half-life (days) |
|---|---|---|
Sulfur-32 | Phosphorus-32 | 14.28 |
Sulfur-33 | Phosphorous-33 | 25.3 |
Titanium-47 | Scandium-47 | 3.35 |
Yttrium-89 | Strontium-89 | 50.52 |
Mercury-199 | Gold-199 | 3.15 |
Zinc-67 | Copper-67 | 2.58 |
Several key therapeutic radioisotopes of current or potential interest can be reactor-produced and primarily include beta-emitting radioisotopes, although some Auger electron emitters and alpha emitters can also be produced in research reactors. The following sections summarize details of the production and chemical purification of several key examples.
5.7 Beta-Particle-Emitting Radionuclides
5.7.1 Arsenic-77
Arsenic-77 (T 1/2 = 1.6 d) is a promising medium-range β−-emitting radionuclide (E βmax = 0.7 MeV) for in vivo radionuclide therapy owing to its favorable nuclear decay characteristics (Neves et al. 2002), decaying by 100 % via electron emission to stable 77Se. The emission of low-energy gamma photons [239 keV (1.6 %), 520 keV (0.5 %)] allows imaging for evaluation of in vivo radiopharmaceutical localization and pharmacokinetics (Jennewein et al. 2006). The 1.6-d half-life of 77As is compatible with the pharmacokinetics of several targeting biomolecules, including monoclonal antibodies (MAbs) (Emran and Phillips 1991) and is amenable for radiochemical synthetic procedures which may require several hours. The chemistry for effective labeling of biologically relevant molecules with arsenic radioisotopes is slowly evolving (Leonard 1991; Emran 1984; Hosain et al. 1982; Jennewein et al. 2003a, b; Zhu et al. 1997; Mirzadeh and Lambrecht 1996). Arsenic forms stable covalent bonds with carbon and sulfur and can substitute phosphorus in certain compounds with minimal alteration of the biological activity of the parent molecules. In addition, the potential use of 77As for endoradiotherapeutic radiopharmaceuticals has also been proposed (Maki and Murakami 1974).
Reactor production of 77As involves the 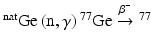 As reaction, where GeO2 or Ge metal targets of natural abundance are irradiated to undergo (n,γ) activation to produce 77Ge (Table 5.2), which subsequently decays by β− emission (t ½ = 11.3 h) to yield 77As (Jennewein et al. 2005; Maki and Murakami 1974). Natural Ge is a mixture of five isotopes, 70Ge, 72Ge, 73Ge, 74Ge, and 76Ge, with relative abundances of 21.23 %, 27.66 %, 7.72 %, 35.94 %, and 7.45 %, respectively (Jennewein et al. 2006). Neutron irradiation of this isotopic mixture results produces a mixture containing 71mGe, 71Ge,70Ga,72Ga,73Ga, 74Ga, 75mGe,75Ge, 76Ga, 77mGe,77Ge, 69mZn, 69Zn,71mZn,71Zn, and 73Zn (Erdtmann 1976). With the exception of 71Ge (T ½ = 11.2 d), the other activated Ge radionuclides are short-lived. The production of radioactive Ga and Zn radioisotopes is negligible owing to the very low neutron cross sections. On the other hand, 24 % of the 77mGe produced (t ½ = 54 s) rapidly decays to 77Ge (t ½ = 11.3 h), while the remainder disintegrates directly to 77As (t ½ = 38.7 h) by β− decay, which finally decays to stable 77Se. During target cooling, most of the short-lived radionuclides decay, which not only facilitates the in-growth of 77As to maximum levels but also reduces the radiation dose to a considerable extent.
As reaction, where GeO2 or Ge metal targets of natural abundance are irradiated to undergo (n,γ) activation to produce 77Ge (Table 5.2), which subsequently decays by β− emission (t ½ = 11.3 h) to yield 77As (Jennewein et al. 2005; Maki and Murakami 1974). Natural Ge is a mixture of five isotopes, 70Ge, 72Ge, 73Ge, 74Ge, and 76Ge, with relative abundances of 21.23 %, 27.66 %, 7.72 %, 35.94 %, and 7.45 %, respectively (Jennewein et al. 2006). Neutron irradiation of this isotopic mixture results produces a mixture containing 71mGe, 71Ge,70Ga,72Ga,73Ga, 74Ga, 75mGe,75Ge, 76Ga, 77mGe,77Ge, 69mZn, 69Zn,71mZn,71Zn, and 73Zn (Erdtmann 1976). With the exception of 71Ge (T ½ = 11.2 d), the other activated Ge radionuclides are short-lived. The production of radioactive Ga and Zn radioisotopes is negligible owing to the very low neutron cross sections. On the other hand, 24 % of the 77mGe produced (t ½ = 54 s) rapidly decays to 77Ge (t ½ = 11.3 h), while the remainder disintegrates directly to 77As (t ½ = 38.7 h) by β− decay, which finally decays to stable 77Se. During target cooling, most of the short-lived radionuclides decay, which not only facilitates the in-growth of 77As to maximum levels but also reduces the radiation dose to a considerable extent.
As reaction, where GeO2 or Ge metal targets of natural abundance are irradiated to undergo (n,γ) activation to produce 77Ge (Table 5.2), which subsequently decays by β− emission (t ½ = 11.3 h) to yield 77As (Jennewein et al. 2005; Maki and Murakami 1974). Natural Ge is a mixture of five isotopes, 70Ge, 72Ge, 73Ge, 74Ge, and 76Ge, with relative abundances of 21.23 %, 27.66 %, 7.72 %, 35.94 %, and 7.45 %, respectively (Jennewein et al. 2006). Neutron irradiation of this isotopic mixture results produces a mixture containing 71mGe, 71Ge,70Ga,72Ga,73Ga, 74Ga, 75mGe,75Ge, 76Ga, 77mGe,77Ge, 69mZn, 69Zn,71mZn,71Zn, and 73Zn (Erdtmann 1976). With the exception of 71Ge (T ½ = 11.2 d), the other activated Ge radionuclides are short-lived. The production of radioactive Ga and Zn radioisotopes is negligible owing to the very low neutron cross sections. On the other hand, 24 % of the 77mGe produced (t ½ = 54 s) rapidly decays to 77Ge (t ½ = 11.3 h), while the remainder disintegrates directly to 77As (t ½ = 38.7 h) by β− decay, which finally decays to stable 77Se. During target cooling, most of the short-lived radionuclides decay, which not only facilitates the in-growth of 77As to maximum levels but also reduces the radiation dose to a considerable extent.The production, purification, and application of 77As has been an active area of research in nuclear medicine and radiochemistry communities and 77As can be cyclotron-/accelerator-produced by the deuteron-induced reaction on enriched 76Ge targets by the 76Ge(d,n) 77As reaction, but this route has not been studied in detail (Jennewein et al. 2005). For processing, 77As can be separated from cyclotron- or reactor-irradiated germanium or germanium oxide targets by methods involving distillation, coprecipitation, solvent extraction, thin layer chromatography, etc. (Jennewein et al. 2005; Maki and Murakami 1974; Chattopadhayay et al. 2007; Bokhari et al. 2009; Mirzadeh and Lambrecht 1996). Four successful preparative-scale separations of 77As from Ge for clinical applications have been reported in the literature (Jennewein et al. 2005; Chattopadhayay et al. 2007; Bokhari et al. 2009; Chakravarty et al. 2011). One method is based on the formation of soluble GeF6 −2 in concentrated hydrofluoric acid and conversion of As to AsI3 by the addition of KI, followed by separation using a polystyrene-based solid-phase extraction system (Jeenewien et al. 2005). The method reported by Chattopadhayay et al. (2007) involves precipitation of GeO2 followed by centrifugation or filtration and subsequent purification using six cycles of solvent extraction (twice with CCl4 and four times with benzene). In an alternative approach, the target is initially dissolved in aqua regia, followed by removal of excess acid by evaporation, reconstitution of the feed in basic media, and passage of the solution through a hydrous zirconium oxide (HZO) column (Bokhari et al. 2009). The Ge is quantitatively retained on HZO, while 77As is present in the effluent. The method adapted by BARC (Chakravarty et al. 2011) consists of a two-step ion-exchange procedure using a polymer-embedded nanocrystalline titania sorbent. The first step involves removal of the bulk Ge from 77As and the second step involves purification and concentration.
5.7.2 Copper-67
Copper-67 (T 1/2 = 2.6 d) is a medium-energy β− emitter with an energy of 0.6 MeVmax (141 keV average), with strong gamma emissions at 91.266 (7 %), 93.31 (16 %), and 184.57 keV (48.7 %) (Junde et al. 2005). Because of the degree of soft tissue penetration, the medium-energy β− particle permits efficient use of 67Cu for effective irradiation and sterilization of cells in solid tumors up to 4 mm in diameter. The range of the 67Cu beta particle in tissue is of the same order as a cell diameter (Qaim 2001). The chemistry of copper is suitable for labeling purposes and precludes the concentrate in sensitive body tissues such as the bone marrow. The 2.6-d half-life is suitable for imaging slow in vivo pharmacokinetics with agents such as monoclonal antibodies and other carrier molecules without appreciable decay loss (DeNardo et al. 1999). The presence of relatively low-energy gamma photos can be effectively detected with Anger cameras [(184 keV 948.7 %), 93.3 (16.1 %)] and is useful for imaging studies (Shen et al. 1996). In vivo, 67Cu in the inorganic form has favorable short-term retention, and both 67Cu and its stable 67Zn decay product are nontoxic since Cu and Zn are essential trace nutrients (Blower et al. 1996). Copper is not a skeletal or organ seeker and 67Cu has a biological half-life of the same order as its radiological half-life (Johnson et al. 1992; Linder and Hazegh-Azam 1996).
Unfortunately, 67Cu cannot be effectively produced in a research reactor in sufficient activity levels to make this route practical. Reactor production of 67Cu via the 67Zn(n, p)67Cu reaction has been reported by bombarding 67Zn with neutrons in high-flux reactors having a high epithermal neutron component; however, the cross-section values are low (Mausner et al. 1998; Johnsen et al. 2015). Production of 67Cu can be much more effectively carried out following using accelerator-based pathways (Katabuchi et al. 2008; Medvedev et al. 2012; Smith et al. 2012; Szelecsényi et al. 2009) and the cyclotron production of 67Cu and different nuclear reactions are discussed and summarized in Table 6.2 in Chap. 6.
5.7.3 Erbium-169
Erbium-169 (T 1/2 = 9.4 d) emits beta particle ([E β(max) = 351 keV (55 %), 342 (45 %)]) with an average soft tissue range of 0.3 mm and is used in targeted therapy as a soft β−-emitting radionuclide, primarily for synovectomy of small joints as described in Chap. 14. Production of 169Er can be conveniently carried out through neutron capture of stable 168Er by the 168Er(n, γ)169Er reaction. Naturally occurring erbium is composed of six stable isotopes, which include 162Er (0.14 %), 164Er (1.61 %),166Er (33.6 %), 167Er (22.95 %),168Er (26.8 %), and 170Er (14.9 %). Neutron irradiation of nat Er not only leads to the coproduction of 165Er and 171Er as impurities but also produces low-SA 169Er. In order to circumvent such drawbacks, isotopically enriched 168Er targets are generally used for 168Er production. Owing to the poor neutron capture cross section of this process (σ = 1.95 barn), however, the activation product obtained is in only low specific activity.
Reactor production and electrochemical purification of 169Er for vivo therapeutic applications have been reported (Chakravarty et al. 2014). Despite having useful radiation emission characteristics, poor specific activity is thus a major impediment that restricts the use of 169Er for radiosynovectomy. However, use of 169Er for the labeling of tumor receptor or antigen-targeting vectors is precluded because of the requirement for much higher specific activities for these applications. 169Er is the most preferred choice for RSV of digital joints such as metacarpophalangeal metatarsophalangeal and digital interphalangeal joints (Karavida and Notopoulos 2010). Erbium-169-labeled silicate/citrate colloid is widely used as in the treatment of refractory painful arthritis (Gumpel et al. 1979; Bouvier et al. 1983; Kahan et al. 2004). Low volumes containing 0.5–1 mCi (20–40 Mbq) with a maximum of 20 mCi (750 Mbq) per administration are used for this application.
5.7.4 Gold-198
Gold-198 (T 1/2 = 2.7 d) has been of interest for many years and decays with emission a β− particle with a maximum energy of 0.96 MeV (99 %) suitable for therapeutic applications. However, the high abundance of high-energy gamma photons (412 keV) (95.6 %) can be a significant disadvantage. Because of the unique properties of gold nanoparticles, in recent years there has been interest in designing and developing 198Au nanoparticles for tumor therapy applications (Chanda et al. 2010). The 198Au is reactor-produced by direct (n,γ) activation by the 197Au(n, γ)198Au reaction which has a very high thermal neutron cross section (σ 26,500 barn). The target used is either gold foil or metal and the neutron-irradiated target is dissolved in aqua regia (3:1 HCl/HNO3) and partially evaporated to yield HAuCl4 in dilute HCl (normally 0.05 M). In order to insure the product is free from trace radionuclide impurities, the AuCl4 − is extracted into organic solvents such as chloroform, dichloromethane, or ethyl acetate as its tetrabutyl ammonium salt, TBA (AuCl4).
5.7.5 Gold-199
Gold-199 [T 1/2 = 3.14 d] also decays by emission of β− particles (E β(max) = 452 keV (6.5 %), 293 keV (72 %), 244 keV (21.5 %)) and gamma photons [(E γ = 494 keV (97 %), 158 keV (36.9 %)]. Although the high gamma abundance is a disadvantage, interest in the potential use of 199Au for therapeutic applications has persisted. Radiolabeling of gold clusters containing 11 gold atoms site selectively to monoclonal antibodies has been reported (Hainfeld 1987; Vaughan and Varley 1988; Hainfeld 1988; Hainfeld et al. 1990). Production of 199Au is performed by an indirect method, involving neutron capture of 198Pt followed by β− decay of the 199Pt (T 1/2 = 30.80 m) to generate 199Au (Table 5.1). The reaction cross section is moderate with a value of 3.6 barn.
The use of natural platinum leads to the concomitant production of 193Pt and 197Pt which results in the production of inactive 197Au, thereby decreasing the 199Au SA. Use of enriched targets is thus necessary to avoid handling high activity levels arising from decay of other Pt radionuclides. The indirect method of production leads to the production of NCA 199Au and the radiochemical separation procedure are well described in the literature (Kolsky and Mausner 1993; Bonardi et al. 2001; Das et al. 1999). Since the 199Au is available NCA, this radionuclide is of interest for radiolabeling peptides and antibodies. However, stabilization of gold by chelation is a challenging task, due to the inertness of this metal.
5.7.6 Holmium-166
Holmium-166 (T 1/2 = 26.83 h) emits β− particles with a maximum energy of 1.85 MeV [E β(max) = 1854 keV (50.0 %), 1774 keV (48.7 %)] and γ rays with energies of 80.6 (6.2 %) and 1379 keV (1.13 %). The 80 keV photon is within the optimal energy range for effective gamma camera imaging. The two β− emissions have mean soft tissue penetration range of 4 mm and a maximum range of 8.7 mm in soft tissues (Marque et al. 2005; Nijsen et al. 1999). Holmium-166 is an excellent radionuclide suited for in vivo therapeutic applications (Dadachova et al. 1997; Hong et al. 2002, 2004; Chakraborty et al. 2001; Chakraborty et al. 2006b; Majali et al. 2001, 2002; Das et al. 2009a, b), although the relatively short half-life may often limit distribution of 166Ho-labeled radiopharmaceuticals to within only short proximity to the production site. Although 166Dy/166Ho radionuclide generator prototypes have been described, these systems are not yet further developed or practical for routine clinical use (see Chap. 7). The most widely utilized 166Ho radiopharmaceutical which has been described for therapy rather than palliation of bone cancer is 166Ho-DOTMP (DOTMP = 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetramethylene-phosphonic acid; Fig. 11.12, Chap. 11), which targets multiple myeloma, a cancer of the plasma white blood cells, that is presented in the bone marrow of these patients (Breits et al. 2006; Rajendren et al. 2002). Utility of 166Ho-EDTMP for bone pain palliation has also been explored (Appelbaum et al. 1992; Bahrami-Samani et al. 2010; Sohaib et al. 2011).
For the treatment of unresectable liver cancer (see Chap. 11), several 166Ho-labeled agents are in various stages of evaluation and include the percutaneous administration of the 166Ho–chitosan complex (PHI) (Rajendren et al. 2002), the 166Ho–oxine–lipiodol complex (Das et al. 2009a, b) and 166Ho–polylactic acid microspheres (Nijsen et al. 1999).
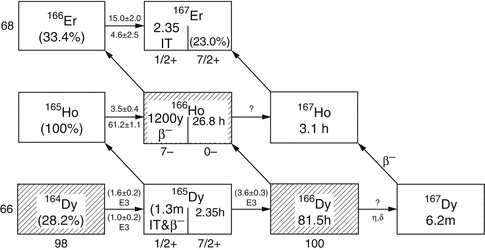
Fig. 5.2
Reactor production of 166Ho
Holmium-166 can be produced by direct neutron irradiation of 165Ho, which is 100 % abundant (Table 5.1) and from decay of reactor produced 166Dy (Fig. 5.2). Since the thermal neutron activation cross section is 66 barn, relatively high activity levels of 166Ho can be produced, but with only moderate specific activity, because only a very small portion (~0.31 %) of target atoms are actually converted to 166Ho at saturation yields (Fig. 5.3).
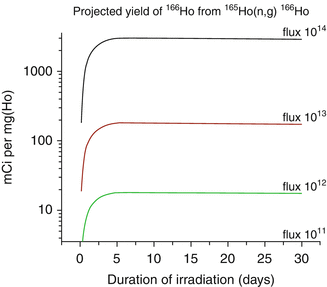
Fig. 5.3
Theoretical production yields of 166Ho from irradiation of 165Ho as a function of thermal neutron flux
Although the modest SA of the 166Ho produced by direct (n,γ) activation is not useful for radiolabeling receptor-specific biomolecules, these 166Ho SA values are sufficient for several applications where much higher radiopharmaceutical masses can be administered, which include bone marrow ablation, therapy of hepatocellular carcinoma, and radiosynovectomy. For targeting low populations of binding sites such as receptor binding, the use of NCA 166Ho is required, and the availability of NCA 166Ho from a 166Dy/166Ho generator would be expected to be of interest for these applications. Potential of 166Dy/166Ho in vivo generator system for therapeutic use have also been explored (Ferro-Flores et al. 2003; 2004). Production of 166Ho through hot atom reactions has also been explored (Nassan et al. 2011; Zeisler and Weber 1998).
For such applications, NCA 166Ho can be produced via an indirect route (Table 5.2) in which an enriched 164Dy target undergoes two sequential neutron captures (2n, γ) to provide 166Dy that subsequently decays by β− emission to the desired 166Ho product by the 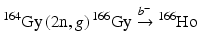 route. This method necessitates availability of efficient methods to perform chemical separation of microscopic levels of 166Ho from macroscopic amounts of 166Dy. The initial neutron capture cross-section values are effectively 2731 barn thermal and 932 barn epithermal to form 165Dy with a half-life of 2.33 h. Although the half-life of this intermediate radionuclide is relatively short, 165Dy has thermal and epithermal neutron capture cross sections of 3600 and 22,000 barn, respectively, and thus a significant percentage of 165Dy atoms undergo second neutron capture reaction to form 166Dy. One milligram of enriched 164Dy irradiated over 155 h at a thermal flux of 4 × 1014 neutrons cm−2 s−1 thermal and an epithermal flux of 1.6 × 1013 neutrons cm−2 s−1 at the MURR has been shown to produce close to the theoretical yield of 1.2 Ci of 166Dy (Ma et al. 1996). Similar production studies at the ORNL HFIR have demonstrated production of 166Dy and separation of 166Ho (Dadchova et al. 1994; 1995; Lahiri et al. 2004). Efforts for separation of NCA 166Ho from 166Dy are described in Chap. 6.
route. This method necessitates availability of efficient methods to perform chemical separation of microscopic levels of 166Ho from macroscopic amounts of 166Dy. The initial neutron capture cross-section values are effectively 2731 barn thermal and 932 barn epithermal to form 165Dy with a half-life of 2.33 h. Although the half-life of this intermediate radionuclide is relatively short, 165Dy has thermal and epithermal neutron capture cross sections of 3600 and 22,000 barn, respectively, and thus a significant percentage of 165Dy atoms undergo second neutron capture reaction to form 166Dy. One milligram of enriched 164Dy irradiated over 155 h at a thermal flux of 4 × 1014 neutrons cm−2 s−1 thermal and an epithermal flux of 1.6 × 1013 neutrons cm−2 s−1 at the MURR has been shown to produce close to the theoretical yield of 1.2 Ci of 166Dy (Ma et al. 1996). Similar production studies at the ORNL HFIR have demonstrated production of 166Dy and separation of 166Ho (Dadchova et al. 1994; 1995; Lahiri et al. 2004). Efforts for separation of NCA 166Ho from 166Dy are described in Chap. 6.
route. This method necessitates availability of efficient methods to perform chemical separation of microscopic levels of 166Ho from macroscopic amounts of 166Dy. The initial neutron capture cross-section values are effectively 2731 barn thermal and 932 barn epithermal to form 165Dy with a half-life of 2.33 h. Although the half-life of this intermediate radionuclide is relatively short, 165Dy has thermal and epithermal neutron capture cross sections of 3600 and 22,000 barn, respectively, and thus a significant percentage of 165Dy atoms undergo second neutron capture reaction to form 166Dy. One milligram of enriched 164Dy irradiated over 155 h at a thermal flux of 4 × 1014 neutrons cm−2 s−1 thermal and an epithermal flux of 1.6 × 1013 neutrons cm−2 s−1 at the MURR has been shown to produce close to the theoretical yield of 1.2 Ci of 166Dy (Ma et al. 1996). Similar production studies at the ORNL HFIR have demonstrated production of 166Dy and separation of 166Ho (Dadchova et al. 1994; 1995; Lahiri et al. 2004). Efforts for separation of NCA 166Ho from 166Dy are described in Chap. 6.5.7.7 Iodine-131
Iodine-131 (T 1/2 = 8.02 d) has been widely used for therapy for many years and has many advantages and has traditionally represented one of the most important and most widely used reactor-produced radionuclides (Ambade et al. 2015). Decay involves emission of medium-energy beta particles [E β(max) = 606 keV (89.9 %), 333 (7.27)] and gamma rays [E γ = 364 keV (81.2 %), 636 (7.27 %)]. The 8.02-day half-life is long enough for uptake and incorporation of radioiodine into thyroglobulin by the follicular thyroid cells, which has been the primary treatment of thyroid cancer. The medium-energy beta particle has a mean path length of about 1 mm in soft issue and is well suited for the treatment of small tumors such as follicular cell carcinoma. The emission of gamma radiation—although of high energy—facilitates imaging, which permits the uptake and localization of low activity levels of 131I to be assessed prior to administration of therapeutic doses. Ready availability of 131I in liquid form at affordable cost permits straightforward dispensing and oral administration. The high-energy gamma radiation of 131I can result in poor images and contributes significantly to the whole-body patient radiation burden without significantly increasing the radiation damage to the target tissue. This factor also adds to the radiation dose to staff and relatives, necessitating in most countries the admission and isolation of patients undergoing radioiodine therapy.
Iodide anions are localized in thyroid cells by active transport and directly incorporated into thyroglobulin from which the thyroid hormones are released. Iodine-131 for the treatment of thyroid disease is utilized for hyperthyroidism or well-differentiated thyroid carcinoma indications. There is also a large and growing body of evidence to support effective clinical use of 131I for management of well-differentiated thyroid cancers (Ambrosetti et al. 2009; Pitoia and Cavallo 2012; Pagano et al. 2004; Reynolds and Robbins 1997; Giovanella 2011). Apart from its utility in the management of thyroid disorder, 131I-MIBG (m-iodobenzylguanidine) is widely used for the treatment of neuroendocrine-originating tumors (Castellani et al. 2000; Bomanji et al. 2003; Gedik et al. 2008; Ezzidin et al. 2012), while 131I-Tositumomab (anti CD20 antibody, Bexaar®) is a registered radiopharmaceutical for the treatment of non-Hodgkin’s lymphoma (Tomblyn 2012; Lewington 2005; Dewaraja et al. 2010; Tsai et al. 2004; Kern 2000). The comparatively long half-life of 131I provides logistical advantage for the radiochemical production and processing/dispensing/shipment of 131I radiopharmaceuticals.
Production of 131I on a large scale can be performed either from 235U(n,f)131I (Siri and Mondino 2005; Nazari et al. 2001; Al-Janabi and Kadem 1990; Tabasi et al. 2005; Wojdowska 2010; Khalafi et al. 2005; Ahmad et al. 1982) or from the 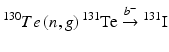 nuclear reactions (Shikata and Amano 1973; Payamara 2011; Chattopadhyay and Saha-Das 2010; El-Absy et al. 2010; Constan 1958; El-Azony et al. 2004; Al-Janabi and Kadem 1990; Elom Achoribo et al. 2012; Alanis and Navarrete 1999; Sorantin and Bildstein 1965). Although the 235U fission route provides HSA 131I, this production method is of limited use because of the requirements for availability of an elaborate complex radiochemical processing technology, sophisticated equipment, a reliably well-trained workforce, and management of high levels of radioactive waste. The production of fission 131I can be sustained only by countries pursuing fission 99Mo production where 131I could be produced as a by-product. Due to the inherent complexities in the production of fission 131I, alternative production technologies using neutron activation of 130Te is widely practiced. The natural abundances of tellurium isotopes and their corresponding thermal neutron (n, γ) cross-section values are summarized in Table 5.4.
nuclear reactions (Shikata and Amano 1973; Payamara 2011; Chattopadhyay and Saha-Das 2010; El-Absy et al. 2010; Constan 1958; El-Azony et al. 2004; Al-Janabi and Kadem 1990; Elom Achoribo et al. 2012; Alanis and Navarrete 1999; Sorantin and Bildstein 1965). Although the 235U fission route provides HSA 131I, this production method is of limited use because of the requirements for availability of an elaborate complex radiochemical processing technology, sophisticated equipment, a reliably well-trained workforce, and management of high levels of radioactive waste. The production of fission 131I can be sustained only by countries pursuing fission 99Mo production where 131I could be produced as a by-product. Due to the inherent complexities in the production of fission 131I, alternative production technologies using neutron activation of 130Te is widely practiced. The natural abundances of tellurium isotopes and their corresponding thermal neutron (n, γ) cross-section values are summarized in Table 5.4.
nuclear reactions (Shikata and Amano 1973; Payamara 2011; Chattopadhyay and Saha-Das 2010; El-Absy et al. 2010; Constan 1958; El-Azony et al. 2004; Al-Janabi and Kadem 1990; Elom Achoribo et al. 2012; Alanis and Navarrete 1999; Sorantin and Bildstein 1965). Although the 235U fission route provides HSA 131I, this production method is of limited use because of the requirements for availability of an elaborate complex radiochemical processing technology, sophisticated equipment, a reliably well-trained workforce, and management of high levels of radioactive waste. The production of fission 131I can be sustained only by countries pursuing fission 99Mo production where 131I could be produced as a by-product. Due to the inherent complexities in the production of fission 131I, alternative production technologies using neutron activation of 130Te is widely practiced. The natural abundances of tellurium isotopes and their corresponding thermal neutron (n, γ) cross-section values are summarized in Table 5.4.Table 5.4
Isotopic abundance of natural tellurium
Tellurium isotope | % Abundance | Nuclear reaction | Thermal neutron cross sections (σ) in barn (b) |
|---|---|---|---|
120Te | 0.089 | 120Te(n, γ)121Te | 2.0 |
120Te(n, γ)121m Te | 0.34 | ||
122Te | 2.46 | 122Te(n, γ)123m Te | 1.1 |
124Te | 4.61 | 124Te(n, γ)125m Te | 0.04 |
126Te | 18.71 | 126Te(n, γ)127Te | 0.9 |
126Te(n, γ)127m Te | 0.135 | ||
128Te | 31.7 | 128Te(n, γ)129Te | 0.14 |
128Te(n, γ)129mTe | 0.015 | ||
130Te | 33.8 | 130Te(n, γ)131Te | 0.2 |
130Te(n, γ)131m Te | 0.04 |
Reactor production of 131I is generally via irradiation of 130Te via the 130Te(n, γ)131Te (t ½ = 25 min) and 130Te(n, γ)131m Te (t ½ = 30 h) nuclear reactions, which have thermal-neutron capture cross sections of 0.2 and 0.04 barn, respectively, and the three decay branches are shown in Figs. 5.4 and 5.5.
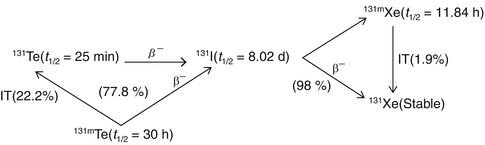
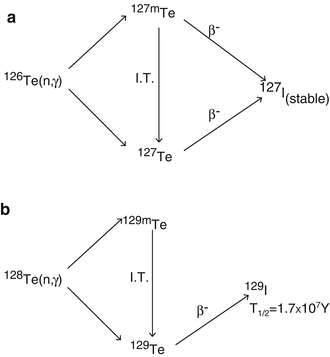
Fig. 5.4
Decay scheme of neutron-irradiated 131Te
Fig. 5.5
Activation reactions and decay mode of (a) 126Te and (b) 128Te formed during the neutron irradiation of natural TeO2
Use of natural tellurium target also results in the production of 127I and 129I as depicted in Table 5.5.
Table 5.5
Other isotopes produced during the neutron irradiation of natural tellurium
Tellurium isotope in target | % Abundance | Isotope produced | T½ | Disintegration products |
|---|---|---|---|---|
120Te | 0.089 | 121Te | 154 days | 121Te → 121Sb(stable) |
121m Te | 16.8 days | |||
122Te | 2.46 | 123m Te | 119.7 days | 123Te (stable) |
124Te | 4.61 | 125m Te | 57.4 days | 125Te (stable) |
126Te | 18.71 | 127Te | 109 days | 127Te → 127I (stable) |
127m Te | 9.35 h | |||
128Te | 31.7 | 129Te | 33.6 days | 129Te → 129I (1.7 × 107 years) |
129mTe | 69.6 min. |
127I is inactive, whereas 129I is long-lived with a half-life of 1.57 × 107 years. The activity levels of 129I formed are very low, due to the long half-life; however, the number of atoms of iodine (127I and 129I) at the end of bombardment (EOB) constitutes about 80 % of the total iodine atoms. Thus the isotopic abundance of 131I will not exceed 20 % at EOB and continues to reduce with progressive decay of 131I. Separation of 131I from irradiated Te is usually carried out following dry and wet chemical distillation method following the reported procedures (Ambade et al. 2015; Ahmad et al. 1982; Beyer and Pimentel-Gonzales 2000). However, dry distillation is preferred due to the significantly lower levels of radioactive liquid waste which are generated, the more rapid processing, and the ability to obtain high radioactive concentrations of the final 131I product.
5.7.8 Lutetium-177
Lutetium-177 (T 1/2 = 6.65 d) represents therapeutic radioisotopes of rapidly increasing importance which decays with emission of low- to medium-energy β− emission [E β(max) = 497 keV (78.6 %), 385 keV (9.1 %)] and photon emissions [E γ = 208 keV (11.0 %), 113 keV (6.4 %)] which permit gamma camera imaging and which can be used as a 3+ lanthanide for facile radiolabeling chemistry. Lutetium-177 is also a very attractive theranostic radionuclide which can be used in diagnostic/therapeutic pairing and exhibits a soft tissue penetration range of several millimeters. The use of 177Lu-labeled peptides and for other applications is described in Chap. 10. The 6.65-day half-life is long enough for preparation of targeted biomolecules which have short or long biological half-lives and allows the products to be distributed for use at clinics distant to the production site. The gamma rays emitted by 177Lu permit imaging of the uptake and biodistribution of the agent both before and during therapy administration. The principal applications of 177Lu currently include treatment of neuroendocrine tumors (Kam et al. 2012; Gulenchyn et al. 2012; Esser et al. 2006), especially for bone pain palliation of metastatic cancer (Bodei et al. 2011; Delpassand et al. 2014; Kwekkeboom et al. 2008; Swärd et al. 2010; van Essen et al. 2010; Zaknun et al. 2013; Chakraborty et al. 2008a, c; Mathe et al. 2010; Liu et al. 2012; Das et al. 2002; Chakraborty et al. 2008a, b, c; Bryan et al. 2009), radiation synovectomy (Chakraborty et al. 2006a, b, c; Abbasi et al. 2011), and non-Hodgkin’s lymphoma (Goldenberg and Sharkey 2006; Michel et al. 2005), and treatment of ovarian (Alvarez et al. 1997) and liver cancer (Chakraborty et al. 2008a, b, c) have also been studied.
Two alternative production routes are available for reactor production of 177Lu (Fig. 5.6) which include the “direct” neutron activation of highly enriched 176Lu targets by the 176Lu (n, γ)177Lu reaction, which can provide relatively high SA 177Lu in medium- to high-flux research reactors due to the high thermal neutron cross section (Chakraborty et al. 2014; Dash et al. 2015a, b). The “indirect” route is based on neutron irradiation of enriched ytterbium targets by the 176Yb(n, γ)177Yb → β− route to NCA 177Lu. The radiochemical processing from the “direct” route involves simple dissolution of the irradiated targets, whereas purification of HSA 177Lu from the “indirect” route involves more elaborate radiochemical processing to separate the microscopic levels of NCA 177Lu from macroscopic ytterbium targets and other radionuclides.
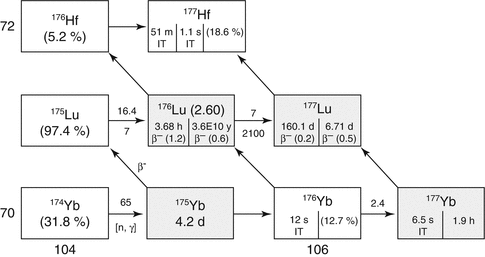
Fig. 5.6
Reactor production of 177Lu
In contrast to production of many radionuclides by the (n, γ) route (see Table 5.1), the direct neutron activation method of 177Lu production is attractive because of the high thermal neutron capture cross-section value for 176Lu (σ = 2090 b). In fact, this is the highest cross section encountered among all the therapeutic radionuclides of current interest reactor-produced by the (n, γ) route. It is possible to produce 177Lu of specific activity of 740–1110 GBq/mg (20–30 Ci/mg) in medium-flux reactors using isotopically enriched 176Lu in which only 20 − 30 % of the 176Lu atoms are being converted to 177Lu. The specific activity of 177Lu can be augmented to 1850–2405 GBq/mg (50–65 Ci/mg) by irradiation in high-flux reactors such as the high-flux isotope reactor (HFIR) at Oak Ridge National Laboratory (ORNL) and at the SM3 reactor in Dimitrovgrad, Russian Federation. Experimental setup for radiochemical processing of irradiated lutetium target by the (n, γ) route is shown in Fig. 5.7.
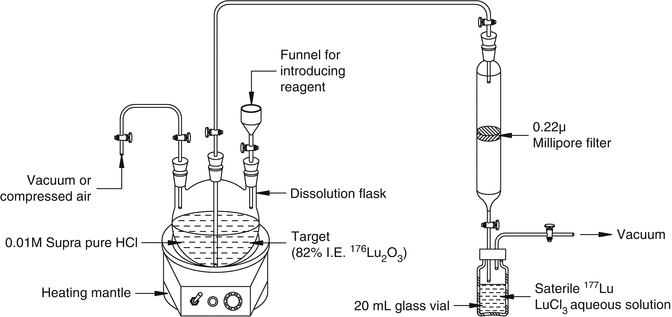
Fig. 5.7
Experimental setup for radiochemical processing of irradiated Lu2O3 target
The direct (n, γ) 177Lu production route also leads to the co-production of low levels of the long-lived 177mLu impurity which has a half-life of 160 days. Although the presence of low activity levels of the 177mLu isomer is not expected to increase the patient radiation burden (Breeman et al. 2003), the presence of this long-lived radiocontaminate in radioactive waste may pose an issue in some countries (Bakker et al. 2006). For this reason, a careful optimization of the time of irradiation may be essential to obtain the highest SA and to also minimize 177mLu contamination levels (Chakraborty et al. 2014; Dash et al. 2015a, b; Pillai et al. 2003). While the use of isotopically enriched 176Lu is a prerequisite for providing177Lu of adequate SA by the “direct” route amenable for radiolabeling peptides and antibodies, use of natural Lu targets is adequate to obtain 177Lu of moderate SA adequate for preparation of several radiopharmaceuticals where high SA is not required for therapeutic application such as bone pain palliation, synovectomy, etc. Use of natural targets (see Table 5.6) does not pose any challenges with respect to radionuclidic impurity.
Table 5.6
Neutron activation products of natural lutetium and ytterbium targets along with the nuclear decay characteristics of the product radionuclides
Element | Target isotope | % Abundance | σ (barn) | Activation product | Mode of decay | T 1/2 | Decay product |
|---|---|---|---|---|---|---|---|
Lu | 175Lu | 97.41 | 16.7 | 176mLu | β−, γ | 3.66 h | 176Hf |
6.6 | 176Lu | β−, γ | 4 × 1010 y | 176Hf | |||
176Lu | 2.59 | 2.8 | 177mLu | β−, γ & IT | 160.4 d | 177Hf (78.6 %) 177Lu (21.4 %) | |
2090 | 177Lu | β−, γ | 6.65 d | 177Hf | |||
Yb | 168Yb | 0.13 | 2300 | 169Yb | EC | 32.02 d | 169Tm |
170Yb | 3.04 | 9.9 | 171Yb | Stable | |||
171Yb | 14.28 | 58.3 | 172Yb | Stable | |||
172Yb | 21.83 | 1.3 | 173Yb | Stable | |||
173Yb | 16.13 | 15.5 | 174Yb | Stable | |||
174Yb | 31.83 | 63 | 175Yb | β−, γ | 4.18 d | 175Lu | |
176Yb | 12.76 | 2.85 | 177Yb | β−, γ | 1.9 h | 177Lu |
During irradiation of natLu, some of the 175Lu atoms are transmuted to 176Lu atoms during neutron irradiation which are subsequently transformed to 176Lu, which enhances the contribution of 177Lu due to double-neutron activation cross section (Fig. 5.8). The contribution of 175Lu to 177Lu production is higher in high-flux reactors since the 177Lu formed from 175Lu will be proportional to the square of the neutron flux (σ2). The tremendous prospects associated with the (n, γ) method of 177Lu production have thus witnessed extensive activity directed toward the establishment of production strategies using a wide spectrum of reactors of varying neutron flux (Mikolajczak et al. 2004; Nir-El 2004; Dvorakova et al. 2008; Zhernosekov et al. 2008; Chinol et al. 2010).
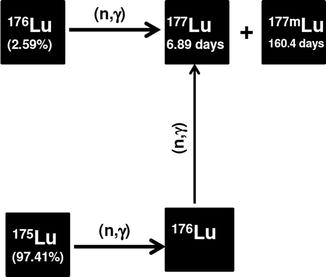
Fig. 5.8
Production of 177Lu using natural Lu
In order to obtain NCA 177Lu, several investigators have described radiochemical separation of NCA 177Lu following neutron irradiation of macroscopic ytterbium targets (Mirzadeh et al. 2004; So et al. 2008; Morcos et al. 2008; Horwitz et al. 2005; Hashimoto et al. 2003; Lebedev et al. 2000; Lahiri et al. 1998; Bilewics et al. 2009; Kumric et al. 2006; Chakraborty et al. 2008b; Knapp et al. 2005; Dash et al. 2015a, b). In this method, an isotopically enriched 176Yb target undergoes (n, γ) transmutation to produce 177Yb which subsequently decays by β−-emission (T ½ = 1.9 h) to yield 177Lu by the 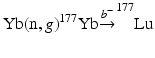 process. The prospects associated with this route of 177Lu production along with the challenge associated with the separation of chemically similar neighboring lanthanides have thus led to a considerable research and innovative detection strategies. At least four successful preparative-scale separations of 177Lu from Yb for clinical applications have been reported in the literature (Lebedev et al. 2000; Bilewicz et al. 2009; Mirzadeh et al. 2004; Chakravarty et al. 2010). The method reported by Lebedev et al. is based on the selective extraction of Yb by sodium amalgam from Cl−/CH3COO− electrolytes taking advantage of the higher solubility of metallic Yb than Lu in mercury. This rather complex and time-consuming process involves eight cementation cycles. The separated 177Lu is reported to contain 10 μg Yb(III) from 200 mg of neutron-irradiated Yb2O3 and must be subjected to an ion exchange process for purification. Subsequently, Bilewicz et al. (2009) have reported a method based on the reduction of Yb(III) to Yb(II) with sodium amalgam followed by selective precipitation of Yb(II) as the sulfate. The 177Lu solution obtained after the precipitation step contained 1 mg Yb(III) from 50 mg of neutron-irradiated Yb2O3 and was hence subjected to purification by an ion exchange process. The method described by Knapp and co-workers consists of a chromatographic extraction method containing LN resin comprised di(2-ethylhexyl)orthophosphoric acid (HDEHP) in which both Lu and Yb can be loaded. The column was eluted with 2 M HCl and the initial elution 170Tm detected prior to Yb elution was presumed to be formed by neutron activation of the low levels of stable 169Tm impurity present in the enriched 176Yb target material. After the removal of 170Tm and Yb, 177Lu was then eluted with 6 M HCl. The specific activity of 177Lu obtained by this method was estimated to be 3.7 TBq (100 Ci)/mg (i.e., 91 % of the 110 Ci/mg theoretical value).
process. The prospects associated with this route of 177Lu production along with the challenge associated with the separation of chemically similar neighboring lanthanides have thus led to a considerable research and innovative detection strategies. At least four successful preparative-scale separations of 177Lu from Yb for clinical applications have been reported in the literature (Lebedev et al. 2000; Bilewicz et al. 2009; Mirzadeh et al. 2004; Chakravarty et al. 2010). The method reported by Lebedev et al. is based on the selective extraction of Yb by sodium amalgam from Cl−/CH3COO− electrolytes taking advantage of the higher solubility of metallic Yb than Lu in mercury. This rather complex and time-consuming process involves eight cementation cycles. The separated 177Lu is reported to contain 10 μg Yb(III) from 200 mg of neutron-irradiated Yb2O3 and must be subjected to an ion exchange process for purification. Subsequently, Bilewicz et al. (2009) have reported a method based on the reduction of Yb(III) to Yb(II) with sodium amalgam followed by selective precipitation of Yb(II) as the sulfate. The 177Lu solution obtained after the precipitation step contained 1 mg Yb(III) from 50 mg of neutron-irradiated Yb2O3 and was hence subjected to purification by an ion exchange process. The method described by Knapp and co-workers consists of a chromatographic extraction method containing LN resin comprised di(2-ethylhexyl)orthophosphoric acid (HDEHP) in which both Lu and Yb can be loaded. The column was eluted with 2 M HCl and the initial elution 170Tm detected prior to Yb elution was presumed to be formed by neutron activation of the low levels of stable 169Tm impurity present in the enriched 176Yb target material. After the removal of 170Tm and Yb, 177Lu was then eluted with 6 M HCl. The specific activity of 177Lu obtained by this method was estimated to be 3.7 TBq (100 Ci)/mg (i.e., 91 % of the 110 Ci/mg theoretical value).
process. The prospects associated with this route of 177Lu production along with the challenge associated with the separation of chemically similar neighboring lanthanides have thus led to a considerable research and innovative detection strategies. At least four successful preparative-scale separations of 177Lu from Yb for clinical applications have been reported in the literature (Lebedev et al. 2000; Bilewicz et al. 2009; Mirzadeh et al. 2004; Chakravarty et al. 2010). The method reported by Lebedev et al. is based on the selective extraction of Yb by sodium amalgam from Cl−/CH3COO− electrolytes taking advantage of the higher solubility of metallic Yb than Lu in mercury. This rather complex and time-consuming process involves eight cementation cycles. The separated 177Lu is reported to contain 10 μg Yb(III) from 200 mg of neutron-irradiated Yb2O3 and must be subjected to an ion exchange process for purification. Subsequently, Bilewicz et al. (2009) have reported a method based on the reduction of Yb(III) to Yb(II) with sodium amalgam followed by selective precipitation of Yb(II) as the sulfate. The 177Lu solution obtained after the precipitation step contained 1 mg Yb(III) from 50 mg of neutron-irradiated Yb2O3 and was hence subjected to purification by an ion exchange process. The method described by Knapp and co-workers consists of a chromatographic extraction method containing LN resin comprised di(2-ethylhexyl)orthophosphoric acid (HDEHP) in which both Lu and Yb can be loaded. The column was eluted with 2 M HCl and the initial elution 170Tm detected prior to Yb elution was presumed to be formed by neutron activation of the low levels of stable 169Tm impurity present in the enriched 176Yb target material. After the removal of 170Tm and Yb, 177Lu was then eluted with 6 M HCl. The specific activity of 177Lu obtained by this method was estimated to be 3.7 TBq (100 Ci)/mg (i.e., 91 % of the 110 Ci/mg theoretical value).The EXC technique was further exploited Horwitz and co-workers (Horwitz et al. 2005) and was found to be successful for the separation of NCA 177Lu from a 300 mg irradiated ytterbium target. The whole separation process can be broadly divided in to three steps: (1) front-end target removal step, (2) primary separation step, and (3) secondary separation step. While the goals of each separation step differ, it basically consists of separation of Yb and Lu using the LN2 resin followed by concentration and acid adjustment of the Lu-rich eluate using Amberchrom® CG-71 resin. In another independent study, a multi-column solid-phase extraction (SPE) chromatography technique using di-(2-ethylhexyl)orthophosphoric acid (HDEHP)-impregnated OASIS-HLB sorbent-based SPE resins (OASIS-HDEHP) was used (Le et al. 2008a, b). The method was successful for the isolation of several hundred mCi of NCA 177Lu using 50 mg Yb target irradiated in a medium neutron flux nuclear reactor (ϕ = 5.1013 n/cm2/sec).
An alternative method (Fig. 5.9) reported by Chakravarty et al. (Chakravarty et al. 2010) consists of an electro-amalgamation approach in which a two-cycle electrolysis procedure was adopted for separation of 18.5 GBq (500 mCi) of 177Lu from 169-177Yb/177Lu mixture in lithium citrate medium. In the first step, the bulk of Yb target is removed, and in the second step, tracer quantities of Yb present in the 177Lu are removed to obtain NCA grade 177Lu.
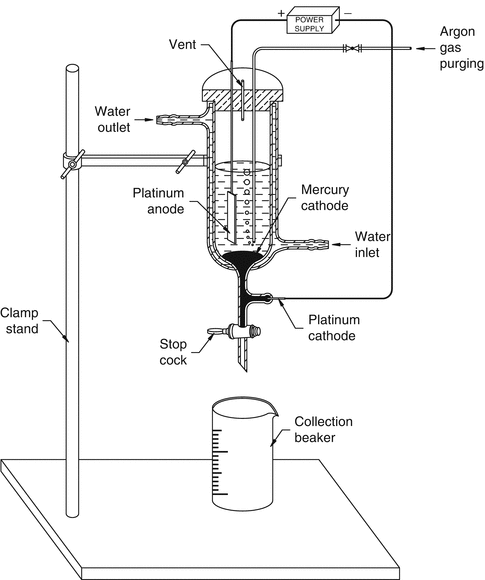
Fig. 5.9
Schematic diagram of the electrochemical setup used for the production of 177Lu using the electro-amalgamation approach
5.7.9 Phosphorous-32
Phosphorous-32 (T 1/2 = 14.26 d) is the first radionuclide used in therapeutic applications more than 50 years ago for the palliation of skeletal pain from bone metastases. Phosporus-32 is a pure β− emitter with a maximum energy of 1.71 MeV and the mean and the maximum particle range in soft tissue are 3 and 8 mm, respectively. The traditional clinical application of 32P has been of the treatment of polycythemia vera and leukemia (Najean and Rain 1997; Tefferi and Silverstein 1998; Meuret et al. 1975; Najean et al. 1996; Tefferi 2012). Phosphorus-32 was first produced in the cyclotron at the University of California in Berkeley, by E. Lawrence in 1936 by irradiating red phosphorus by the 31P(d,p)32P [σ = 0.18 × 10−27cm2] nuclear reaction (Lawrence and Cooksey 1936). Subsequently, when the first nuclear reactor (Graphite Reactor) became available at the Oak Ridge National Laboratory, production shifted to the reactor route, since 32P can be readily produced by either the 32S(n,p)32P or 31P(n, γ)32P nuclear reactions. Only relatively low SA 32P can be produced by the neutron irradiation of 31P (100 % abundance, σ = 0.172 barn) by the 31P(n, γ)32P route but could be used for the preparation of formulations for bone pain palliation (Vimalnath et al. 2013; 2014a, b). This production route requires only very simple chemical treatment after neutron irradiation, but the low SA restricts the useful therapeutic applications of 32P produced by this method.
For these reasons, 32P obtained by separation from the sulfur target irradiated in a fast neutron flux by the 32S(n,p)32P reaction (σ = 0.068b barn for fast neutrons) is currently used for the majority of research and clinical therapeutic applications. The 32S(n,p)32P nuclear reaction involves ejection of a charged particle which must overcome the Coulomb barrier and this reaction is thus favored with fast neutrons. However, the cross section of the 32S(n,p)32P reaction is only 0.068 barn for fast neutrons, and therefore, several hundred grams of sulfur are required for irradiation in order to provide a few hundred mCi of the 32P product. Even though the cross-section values for the 32S(n,p)32P nuclear reaction are much lower than for the 3lP(n, γ)32P route, the irradiation of 32S targets is still of interest and utility because of the production of higher-SA 32P. The isotopic composition of natS consists of 32S (95.02 %), 33S (0.75 %), 34S (4.21 %), and 36S (0.02 %). It is imperative to consider all the radionuclides produced both by thermal and fast neutrons when sulfur undergoes reactor irradiation; Table 5.7 summarizes the nuclear reactions which occur when natural sulfur undergoes irradiation with thermal and fast neutrons in a nuclear reactor.
Table 5.7
Radionuclides produced by irradiation of natural sulfur with thermal and fast neutrons
Isotope of S | Isotopic abundance (%) | Activation products formed | |||
|---|---|---|---|---|---|
Fast neutrons irradiation | Thermal neutrons irradiation | ||||
Nuclear reaction | Half-life | Nuclear reaction | Half-life | ||
32S | 95.02 | 32S(n,p)32P | 14.26 d | 32S(n, γ)33S | Stable |
33S | 0.75 | 33S(n,p)33P | 25.3 d | 33S(n, γ)34S | Stable |
34S | 4.21 | 34S(n,p)34P | 14.4 s | 34S(n, γ)35S | 87.2 d |
36S | 0.02 | 36S(n,p)36P | 5.9 s | 36S(n, γ)37S | 5.05 m |
From an analysis of the possible radionuclides formed by neutron irradiation of natural sulfur, only 35S (T 1/2 = 87.2 d) and 33P (T 1/2 = 25.3 d) are of concern and could be radiochemical impurities of the final 32P product (T 1/2 = 14.28 d). The 35S levels contaminating the desired 32P product are of concern, since they will be carried through the chemical separation process. The contribution of the longer-lived 33P is a point of concern since separation from 32P is not possible by traditional methods and it is not feasible to reduce the 33P levels by cooling because of the longer half-life than 32P. It has been experimentally observed that the 32P and 33P activity ratio produced in the same irradiation conditions is about 7000, because of the much lower isotopic abundance and low fast neutron cross section of the 33S target as well as longer half-life of 33P.
The bulk separation of only a few micrograms of 32P from the bulk of 32S must be conducted, and the 32P SA obtained can be reduced by introduction of inactive phosphorous impurities during radiochemical processing. Due to the low neutron capture reaction cross section, significant large target irradiation volumes are required in the reactor for long periods of time in order to produce 32P. The separation and purification of 32P from irradiated S is usually carried out following wet chemical extraction and dry distillation methods. The wet extraction method consists of dissolution of irradiated powdered sulfur in boiling water in the presence of strong and weak acids to extract the phosphorus nuclide from the sulfur target using 2-octanol (Samsahl 1958; Razbash et al. 1991). One of the major impediments of this wet chemical extraction method is the dependence of extraction yield on the target sulfur particle size which is significantly decreased when the target is melted or solidified due to the exothermal heat generated during neutron irradiation. Additionally, the use of mineral acid is a major deterrent since its use introduces impurities and leaves solid waste behind which not only complicates the extraction process but also requires additional purification steps to achieve the required high chemical and radiochemical purity. The requirement of multistage processes results in poor recovery yields. Because of these drawbacks, the wet chemical extraction method is not often used for 32P production.
The most widely used methods for the separation of 32P from neutron-irradiated sulfur thus involve distillation methods, for instance, at 500 ° C in a nitrogen atmosphere in order to preclude the possibility of fire. Vacuum distillation of sulfur at 180–200 °C, which is lower than the sulfur ignition point, is conducted under a pressure of 1–10 mmHg (Gharemano et al. 1983; Yeh 1962). The distillation method has the advantage that high-purity products can be obtained since no reagents are added during the separation of 32P from neutron-irradiated sulfur. The distillation method usually consists of a vacuum system, gas-feeding apparatus, and distillation assembly (Arrol 1953), in which pressure and temperature are optimally controlled (Gharemano et al. 1983; Yeh 1962; Arrol 1953; Alanis and Navarrete 2007; Karelin et al. 2000). Experimental setup for the radiochemical processing of irradiated elemental sulfur target is shown in Fig. 5.10.
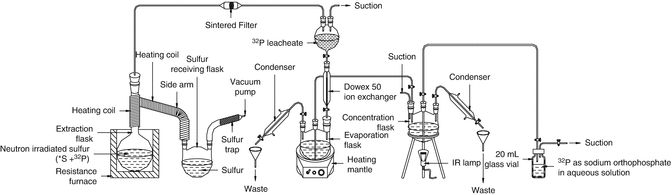
Fig. 5.10
Experimental setup for the radiochemical processing of irradiated elemental sulfur target
5.7.10 Praseodymium-143
Praseodymium-143 (T 1/2 = 13.57 d) is a long-lived medium-energy pure β−-emitting [E β(max) = 934 keV (100 %)] radionuclide useful for therapy. Although reactor production of 143Pr can be performed by the 141Pr(n, γ)142Pr(n, γ)143Pr double-neutron capture process, from this method the activated product consists of a 142Pr and 143Pr mixture, the proportion of which will depend on the irradiation duration and length of the post-irradiation decay period. For this reason, the indirect 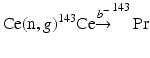 reactor production route is preferred. A target cooling period of a few days is required to insure decay of the short-lived 143Ce (T ½ = 33.04 h), which decays to 143Pr. This production route provides NCA 143Pr with high radionuclidic purity. Naturally occurring Ce is composed of 136Ce (0.185 %), 138Ce (0.251 %), 140Ce (88.45 %), and 142Ce (11.114 %). Neutron activation of natural CeO2 would therefore lead to the formation of l43Ce along with l39Ce, 141Ce, and 143Ce as radionuclidic impurities. In order to reduce the radioactive burden from the irradiated target, use of enriched 142Ce target is mandatory. The reaction cross section has a moderate value of 0.95 barn, which, coupled with the long half-life, not only necessitates long irradiation periods but also requires a high neutron flux irradiation to produce sufficient activity levels of 143Pr (Vimalnath et al. 2005).
reactor production route is preferred. A target cooling period of a few days is required to insure decay of the short-lived 143Ce (T ½ = 33.04 h), which decays to 143Pr. This production route provides NCA 143Pr with high radionuclidic purity. Naturally occurring Ce is composed of 136Ce (0.185 %), 138Ce (0.251 %), 140Ce (88.45 %), and 142Ce (11.114 %). Neutron activation of natural CeO2 would therefore lead to the formation of l43Ce along with l39Ce, 141Ce, and 143Ce as radionuclidic impurities. In order to reduce the radioactive burden from the irradiated target, use of enriched 142Ce target is mandatory. The reaction cross section has a moderate value of 0.95 barn, which, coupled with the long half-life, not only necessitates long irradiation periods but also requires a high neutron flux irradiation to produce sufficient activity levels of 143Pr (Vimalnath et al. 2005).
reactor production route is preferred. A target cooling period of a few days is required to insure decay of the short-lived 143Ce (T ½ = 33.04 h), which decays to 143Pr. This production route provides NCA 143Pr with high radionuclidic purity. Naturally occurring Ce is composed of 136Ce (0.185 %), 138Ce (0.251 %), 140Ce (88.45 %), and 142Ce (11.114 %). Neutron activation of natural CeO2 would therefore lead to the formation of l43Ce along with l39Ce, 141Ce, and 143Ce as radionuclidic impurities. In order to reduce the radioactive burden from the irradiated target, use of enriched 142Ce target is mandatory. The reaction cross section has a moderate value of 0.95 barn, which, coupled with the long half-life, not only necessitates long irradiation periods but also requires a high neutron flux irradiation to produce sufficient activity levels of 143Pr (Vimalnath et al. 2005).Related posts:
 Radionuclide Synovectomy: Treatment of Inflammation of the Synovial Joints
Radionuclide Synovectomy: Treatment of Inflammation of the Synovial Joints
 Introduction: Radiopharmaceuticals Play an Important Role in Both Diagnostic and Therapeutic Nuclear Medicine
Introduction: Radiopharmaceuticals Play an Important Role in Both Diagnostic and Therapeutic Nuclear Medicine
 Inhibition of Arterial Restenosis Following Balloon Angioplasty
Inhibition of Arterial Restenosis Following Balloon Angioplasty
 Therapeutic Radiopharmaceuticals for Treatment of Primary and Metastatic Hepatic Cancer
Therapeutic Radiopharmaceuticals for Treatment of Primary and Metastatic Hepatic Cancer
 Radioimmunotherapy (RIT)
Radioimmunotherapy (RIT)
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


