Sarah J. Vinnicombe, Norbert Avril, Rodney H. Reznek
Reticuloendothelial Disorders
Lymphoma
The lymphomas are complex, but may be divided into two broad groups: Hodgkin’s lymphoma (HL) and non-Hodgkin’s lymphoma (NHL). The overall incidence of NHL has increased steadily since 1960, with age-adjusted incidence rates being highest in more developed countries. It is estimated that there will be 70,000 new cases in 2012, an age-adjusted incidence of 19 per 100,000 persons per year, representing a 73% increase since the early 1970s.1 This is due in part to secondary lymphoma arising in the setting of acquired immune deficiency syndrome (AIDS), though the incidence had started to increase before the AIDS epidemic. In the corresponding period, the incidence of HL has remained relatively steady at around 3 per 100,000.
Epidemiology
Age
HL has a peak incidence in the 20–30-year age group, with a second peak in the elderly population. The incidence of NHL increases exponentially with age after 20 years. The subtypes of lymphoma encountered differ in frequency between adult and paediatric groups, with a strong bias towards precursor B- and T-lymphoblastic lymphoma and Burkitt’s lymphoma (BL) in childhood. Lymphomas with less typical age distribution include mediastinal large B-cell lymphoma, which has a peak incidence between 25 and 35 years, and mantle cell lymphoma, which is more common in those over 60 years.
Infectious Agents
Oncogenic lymphotrophic viruses have been implicated in many types of NHL. The single most important agent in this regard is Epstein–Barr virus (EBV). The EBV genome was first detected in cultured African Burkitt’s lymphoma cells and is present in over 90% of such cases. EBV is important as a trigger for lymphoproliferations/lymphomas occurring in congenital immunodeficiencies, immunosuppressed organ transplant recipients, patients receiving maintenance chemotherapy and patients receiving combined immunosuppressive therapy for collagen disorders. EBV is also found in HL (mostly the mixed cellularity type); patients who have had infectious mononucleosis are at increased risk of HL. The retrovirus human lymphotropic virus type 1 (HTLV–1) is implicated in the causation of adult T-cell lymphoma, which is endemic in certain areas of East Africa, the Caribbean, southwest Japan and New Guinea. Human herpesvirus 8 (HHV–8) has been implicated as a cause of primary effusion lymphoma, a rare type of large cell lymphoma confined to serous-lined body cavities, which occurs with highest frequency in the HIV-positive population. Bacterial overgrowth can also promote lymphomagenesis. In gastric lymphoma of mucosa-associated lymphoid tissue (MALT) type, Helicobacter pylori infection has been shown to be necessary for the development and early proliferation of the lymphoma.
Immunosuppression
A variety of NHL is associated with pre-existing immunosuppression. The degree of immunosuppression is important in determining the lymphoma type that may emerge. In organ-specific autoimmune diseases, such as Hashimoto’s thyroiditis and Sjögren’s syndrome, extranodal marginal zone lymphomas of MALT type can arise within the affected organ. In severe immunodeficiency states, such as the congenital immunodeficiencies, AIDS and after organ transplantation, the lymphomas are very often EBV-driven large B-cell lymphomas. Infection with human immunodeficiency virus (HIV) explains much of the massive increase in the incidence of NHL in the past two decades. In the setting of systemic collagen diseases, there is an increase in haematological malignancy and patients receiving immunosuppressive therapy for these conditions are at still greater risk. The types of haematological malignancy that arise are quite varied, but there is a slight excess of myeloma and small B-lymphocytic lymphoma.
Genetic Factors
It is known that the risk of developing haematological malignancy is increased in patients with a family history of disease. This increased risk does not extend to the histological type or lineage of the tumours in question, such that one family member may have HL whereas a relative may have NHL or myeloid leukaemia.
Gender and Race
There is a slight predominance of NHL and HL in men (1.1–1.4 to 1). The incidence of NHL and HL varies by race, with higher frequencies in whites than blacks or Asians. Certain NHL types cluster according to race: for example, the natural killer (NK) T-cell lymphomas are most frequently encountered in oriental populations.
Histopathological Classification
Hodgkin’s Lymphoma
Biological studies have shown that HL is a true lymphoma. The defining malignant cell of HL is the Reed–Sternberg cell, a large, binucleated blast cell. Mononuclear counterparts are called Hodgkin’s cells. The Reed–Sternberg cells and their variants form a minority population within any involved lymph node. The balance is made up of reactive non-neoplastic T cells, histiocytes, plasma cells, eosinophils and fibroblasts, varying in proportion according to the histological subtype. Since the 1960s, so-called classical HL has been subclassified into four histological types, indicated below along with their relative frequencies in Western populations:
In the nodular sclerosing type, substantial fibroblastic activity results in segmentation of involved nodes into cellular nodules separated by thick bands of collagen. It typically presents as a bulky mediastinal mass. In mixed cellularity HL, classical Reed–Sternberg cells are mixed with sheets of inflammatory elements. It is commoner in developing countries and in patients with HIV infection, as is the rare lymphocyte-depleted HL. Both have an aggressive clinical course. All four classical subtypes share the same immunophenotype and together constitute 95% of all cases of HL. A second distinct entity is nodular lymphocyte predominant HL, which was probably misdiagnosed as lymphocyte-rich classical HL in the past. It has a different morphology, immunophenotype and clinical course from classical HL; latent EBV infection is not a feature.
Non-Hodgkin’s Lymphoma
Many of the difficulties that beset early taxonomists in the classification of NHL have been overcome with improved immunological and molecular methods of diagnosis. The Revised European–American Lymphoma (REAL) classification in 1994 depended on a triad of morphology, immunophenotype and molecular methods as well as clinical features for defining disease entities,2 differentiating it from earlier morphologically based classifications. The scheme forms the backbone of the World Health Organisation (WHO) classification of tumours of haematopoietic and lymphoid tissues.3 A summary of the WHO classification (4th edition) is given in Table 70-1. The WHO classification stratifies neoplasms by lineage into clinically distinct disease entities and is a real advance in the ability to identify disease accurately and consistently. Further, it can be refined so as to improve patient management.4 For example, it has recently been shown that gene expression profiling in diffuse large B-cell lymphoma (DLBCL) enables recognition of discrete subsets (germinal centre B-cell type and activated B-cell type) which have independent prognostic significance, and this has been included in the 4th edition of the classification.3 Other additions include paediatric follicular lymphoma, primary DLBCL of the central nervous system (PCNSL), and two so-called ‘grey zone’ lymphomas: B-cell lymphoma with features intermediate between DLBCL and classical HL, and B-cell lymphoma with features intermediate between DLBCL and BL.
TABLE 70-1
WHO Classification of Lymphoid Neoplasms
| Neoplasm | Percentage of NHL* |
| B-CELL NEOPLASMS | 85.0 |
| Precursor B-cell neoplasms | |
| Precursor B-lymphoblastic leukaemia/lymphoma | |
| Mature B-cell neoplasms | |
| CLL/small lymphocytic lymphoma | 6.7 |
| B-cell prolymphocytic leukaemia | |
| Lymphoplasmacytic (lymphoplasmacytoid) lymphoma | 1.2 |
| Splenic marginal zone lymphoma | <1.0 |
| Hairy cell leukaemia | |
| Plasma cell myeloma | |
| Solitary plasmacytoma of bone | |
| Extraosseous plasmacytoma | |
| Extranodal marginal zone B-cell lymphoma of mucosa-associated lymphoid tissue (MALT) | 7.6 |
| Nodal marginal zone lymphoma | 1.8 |
| Follicular lymphoma | 22.0 |
| Mantle cell lymphoma | 6.0 |
| Diffuse large B-cell lymphoma (BCL) | 32.0 |
| Mediastinal (thymic) | 2.4 |
| Intravascular | |
| Primary effusion lymphoma | |
| Burkitt’s lymphoma/leukaemia | 1.0 |
| B-CELL PROLIFERATIONS OF UNCERTAIN | |
| MALIGNANT POTENTIAL | |
| Lymphomatoid granulomatosis | |
| Post-transplant lymphoproliferative disorder, polymorphic | |
| T-CELL AND NK-CELL NEOPLASMS | 14.0 |
| Precursor T-cell neoplasms | |
| Precursor T-lymphoblastic lymphoma/leukaemia | 1.7 |
| Blastic NK-cell lymphoma | |
| Mature T-cell and NK-cell neoplasms | |
| T-cell prolymphocytic leukaemia | |
| T-cell large granular lymphocytic leukaemia | |
| Aggressive NK-cell leukaemia | |
| Adult T-cell leukaemia/lymphoma | 2.4 |
| Extranodal NK-/T-cell lymphoma, nasal type | <1.0 |
| Enteropathy-type T-cell lymphoma | |
| Hepatosplenic T-cell lymphoma | <0.001 |
| Subcutaneous panniculitis-like | 1.0 |
| T-cell lymphoma | |
| Mycosis fungoides | |
| Sézary syndrome | 1.2 |
| Primary cutaneous anaplastic large cell lymphoma | |
| Peripheral T-cell lymphoma, unspecified | |
| Angioimmunoblastic T-cell lymphoma | 7.0 |
| Anaplastic large cell lymphoma | |
| T-CELL PROLIFERATIONS OF UNCERTAIN MALIGNANT POTENTIAL | |
| Lymphoid papulosis | |
| HODGKIN’S LYMPHOMA | |
| Nodular lymphocyte-predominant HL | |
| Classical HL | |
| Nodular sclerosis classical HL | |
| Lymphocyte-rich classical HL | |
| Mixed cellularity classical HL | |
| Lymphocyte-depleted classical HL |
* Approximate frequency—refers to lymph node biopsies in adults, with 1% accounting for the very rare entities.
Staging, Investigation and Management
Hodgkin’s Lymphoma
Clinical Features and Staging
Most patients present with lymph node enlargement, most often in the cervical chains. Up to 40% have B symptoms (fever, drenching night sweats and weight loss of more than 10% of the person’s bodyweight). Other constitutional symptoms can occur, such as pruritus, fatigue, anorexia and rarely alcohol-induced pain at the site of enlarged lymph nodes. Clinical examination usually reveals lymphadenopathy, most commonly in the neck. Axillary nodal enlargement occurs in up to 20% and inguinal disease in up to 15%, although exclusive infradiaphragmatic nodal disease is seen in up to 10% of patients at presentation. Splenomegaly may be evident on clinical examination in up to 30%.
Tissue biopsy is essential to make the diagnosis. Though a diagnosis of lymphoma may be made from a cutting needle biopsy, surgical excision biopsy of an entire node is preferable, so that the architecture of the node can be evaluated. Investigations will comprise a blood count and erythrocyte sedimentation rate (ESR), together with liver biochemistry, renal function and serum urate. A staging computed tomography (CT) or a diagnostic positron emission tomography (PET)/CT imaging with 2-[F-18]fluoro-2-deoxy-d-glucose (FDG) is mandatory and may show involvement of intrathoracic, abdominal or pelvic lymph nodes. The so-called Cotswold’s modification5 of the original Ann Arbor staging classification (Table 70-2) was designed to take into account prognostic factors such as the volume of lymph node masses as identified with CT. A bone marrow aspirate and trephine biopsy should be performed in all patients except those with stage I disease, in whom the likelihood of bone marrow infiltration is negligible.
TABLE 70-2
Cotswold’s Modification of the Ann Arbor Staging Classification of Hodgkin’s Disease
| Stage | Classification |
| I | Involvement of a single lymph node region (I) or a single extralymphatic organ or site (IE) |
| II | Involvement of two or more lymph node regions on the same side of the diaphragm (II) or one or more lymph node regions plus an extralymphatic site (IIE) |
| III | Involvement of lymph node regions on both sides of the diaphragm (III) (the spleen is included in stage III) subdivided into: |
| IV | Involvement of one or more extralymphatic organs, e.g. lung, liver, bone, bone marrow, with or without lymph node involvement |
| Additional qualifiers denote the following: |

Prognosis and Treatment
For HL a poorer prognosis is noted with:
Treatment is almost invariably given with curative intent. The choice of treatment will depend predominantly on stage, and the presence/absence of adverse prognostic factors. HL is highly radiosensitive, and until recently many patients were treated with ‘mantle’ radiotherapy, which encompassed the cervical nodal chains, the axillae and the mediastinum down to the level of T10. However, there has been a steady trend towards the avoidance of radiotherapy in young patients because of the significant increase in secondary cancers, notably of the thyroid and breast (areas included in the radiotherapy field), and death through coronary artery disease.
Localised Disease (Stages IA and IIA).
The majority of patients with early-stage favourable disease (non-bulky) are treated with combination chemotherapy. The use of interim FDG/PET imaging may allow escalation or de-escalation of therapy, with the goal of avoiding radiotherapy in patients who have a good response to combination chemotherapy and who are therefore in a very good prognostic group.
Advanced Disease (Stages IIB, IIIA/B and IVA/B).
Patients presenting with a large mediastinal mass (i.e. a mass greater than 10 cm in diameter at CT) are generally treated with more intense chemotherapy initially, so as to shrink the mass. Consolidative radiotherapy may then be given. For advanced-stage disease, treatment comprises extensive combination chemotherapy, with or without subsequent consolidatory radiotherapy to sites of ‘bulky’ disease.
Failure to achieve an initial complete or almost complete response to first-line treatment and recurrence in the first year are both associated with a poor prognosis. Patients who develop recurrent HL more than once will generally ultimately die of the disease. By comparison, patients who develop recurrent HL some years after receiving treatment for localised disease can be given further combination chemotherapy and still be cured.
Non-Hodgkin’s Lymphoma
Accurate diagnosis requires adequate tissue biopsy and an experienced histopathologist. Some lesions, for example, retroperitoneal, mediastinal and mesenteric masses, may be amenable to ultrasound or CT-guided core-needle biopsy, which may safely yield adequate tissue for histological diagnosis and immunophenotyping6 but, as with HL, an entire lymph node is preferable for diagnosis.
Clinical Features and Staging
Most patients present with painless lymph node enlargement, but B symptoms are less frequent compared to HL, occurring in approximately 20%. In contradistinction to HL, the histological subtype of NHL is the major determinant of treatment rather than the stage. Nonetheless the stage of the disease has strong prognostic significance, a more advanced stage being associated with a significantly worse prognosis.7 As with HL, the modified Ann Arbor staging system is generally used. Around 80% of patients will have advanced disease (stage III or IV) at presentation, so all newly diagnosed patients should undergo detailed physical examination, including examination of the fauces and testes. As with HL, CT or FDG-PET/CT of the neck, chest, abdomen and pelvis is mandatory, together with a bone marrow aspirate and trephine biopsy, owing to the propensity of most NHL to infiltrate the bone marrow at presentation. Depending on the pattern of symptoms, other radiological investigations such as MRI may be indicated, especially for central nervous system lymphoma.
Prognosis and Treatment
The prognosis of NHL varies tremendously, depending upon the histological subtype. In order to evaluate therapies better and to choose the most appropriate treatment for a given patient, the International Prognostic Index (IPI) was developed.7 The IPI is strictly speaking only applicable to aggressive lymphomas such as diffuse large B-cell lymphoma (DLBCL). Five factors were statistically associated with significantly inferior overall survival:
A similar prognostic index (FLIPI) has been developed for more indolent follicular lymphoma (FL), where the important factors are considered to be:
A recent modification (FLIPI 2) includes elevated serum β2-microglobulin and longest diameter of the largest involved lymph node over 6 cm.8
The histological subtype determines not only the type of treatment but also when treatment should start. For asymptomatic patients with FL, surveillance alone may be appropriate until symptoms develop or transformation to a more aggressive DLBCL occurs. On the other hand, patients with relatively localised DLBCL require treatment with multi-agent anthracycline-containing chemotherapy immediately. Standard treatment for DLBCL and higher-grade FL comprises cyclophosphamide, doxorubicin, vincristine and prednisone (CHOP) combined with rituximab, a chimeric monoclonal antibody against the CD20 receptor, expressed by over 95% of B-cell NHL (CHOP-R). Radiotherapy alone is considered for the small proportion of patients with stage I disease and no adverse factors, in whom surgical excision alone is considered inappropriate.
Lymph Node Disease in Lymphoma
In HL, lymph node involvement is usually the only manifestation of disease, whereas in NHL nodal disease is frequently associated with extranodal involvement. There are differences in the patterns of lymph node involvement in HL and NHL at presentation. Lymph nodes tend to be larger in NHL than HL; indeed, in nodular sclerosing and lymphocyte-depleted HL, nodal enlargement may be minimal. Typically, involved nodes tend to displace adjacent structures rather than invade them, except in the case of primary mediastinal large B-cell lymphoma (PMBL), which is characterised by local invasion of adjacent structures.
Imaging Nodal Disease
At present, size is the only criterion by which lymph nodes demonstrated on CT or MRI are considered to be involved, though clustering of multiple small lymph nodes, for example within the anterior mediastinum or the mesentery, is suggestive. A maximum short-axis diameter of 10 mm is taken to be the upper limit of normal, depending upon the exact site within the neck, thorax, abdomen, or pelvis. There are exceptions: normal neck jugulodigastric node can measure up to 13 mm short-axis diameter; nodes in the gastrohepatic ligament and porta hepatis are considered abnormal if they measure more than 8 mm in diameter; retrocrural nodes greater than 6 mm are taken as enlarged;9 and in the pelvis the upper limit of normal is 8 mm.10 Lymph nodes at some sites, such as the splenic hilum, presacral and perirectal areas, are not usually visualised on cross-sectional imaging and, whenever demonstrated, are likely to be abnormal.
Enlarged lymph nodes in both HL and NHL are usually homogeneous, of soft-tissue density on CT. Mild or moderate uniform enhancement occurs after intravenous injection of contrast medium. Calcification is uncommon but may be seen on post-treatment images. Necrosis is rarely seen in large nodal masses in both HL (particularly nodular sclerosing HL) and aggressive NHL, more frequently after treatment. On MRI, involved lymph nodes have low-to-intermediate signal intensity on T1-weighted images, and they may have very high signal intensity on fat-suppressed T2-weighted and short tau inversion recovery (STIR) sequences. Though the signal intensity of involved nodes and the presence of necrosis do not appear to have much prognostic significance, there is some evidence that within large lymphomatous masses, heterogeneous T2 signal at magnetic resonance imaging (MRI), or heterogeneous enhancement at CT, is associated with a worse outcome.
Choice of Imaging Technique
CT has been the technique of choice for the staging and follow-up of lymphoma for some time, and enables localisation of the most appropriate lesion for consideration of percutaneous image-guided biopsy. Ultrasound has no value in whole-body staging. Ultrasonographic appearances of lymphomatous nodal disease are non-specific, though the pattern of vascular perfusion as demonstrated by power Doppler interrogation may suggest the diagnosis, lymphomatous nodes having rich central and peripheral perfusion. The main value of ultrasound in lymphoma lies in confirming the nature of a palpable mass and assessing the major viscera. The accuracy of MRI in detecting lymph node involvement is equal to that of CT (and is better in some areas such as the supraclavicular fossa and within the pelvis), but it has no particular advantage over CT in this respect and its role is essentially adjunctive, to solve problems or monitor response to treatment. Recent advances in MRI technology (high field strength magnets and parallel imaging) have enabled MRI to be used for whole-body staging: the role of whole-body diffusion-weighted imaging in staging and response assessment is a field of active research.11 Major advantages in patients with HL in particular (who are often young) include the lack of ionising radiation.
Detection of disease in normal-sized nodes is not possible with cross-sectional imaging, nor is it possible to differentiate between nodal enlargement secondary to lymphoma or reactive hyperplasia. Functional imaging is often able to make this distinction. The superior diagnostic accuracy and more favourable imaging characteristics of PET with FDG and, latterly, PET/CT, has resulted in a dramatic decline in the use of gallium-67 scintigraphy, which no longer has a role as an isolated tool in the staging of lymphoma.
Numerous studies have shown that FDG-PET is at least as accurate as CT in the depiction of nodal and extranodal disease12,13 and more sensitive than Ga-67 scintigraphy. It results in clinically significant upstaging in up to 10–20% of patients compared to CT, which may result in changes in therapy, particularly in HL.14 Most NHLs are FDG-avid, though false-negative studies can occur with low-grade lymphomas such as small lymphocytic lymphoma, cutaneous lymphoma, some peripheral T-cell lymphomas and MALT types. For these subtypes, contrast-enhanced CT remains the standard of care. The development of PET/CT with accurate co-registration means that both morphological and functional abnormalities can be assessed simultaneously with improved diagnostic performance of FDG-PET as a result.15 It is important to recognise that lymphomatous involvement of certain organs can be very difficult to recognise with FDG-PET/CT, because of physiological uptake—for example, in the stomach and central nervous system.16 Debate continues as to whether it is necessary to carry out a full diagnostic CT imaging as part of the PET/CT study and often a low-dose CT, for the purposes of attenuation correction and anatomical correlation, is sufficient.17,18
Neck
Between 60 and 80% of patients with HL present with cervical lymphadenopathy. The spread of the disease is most frequently to contiguous nodal groups, with involvement of the internal jugular chain and spread to other deep lymphatic chains in the neck. Patients with supraclavicular or bilateral neck adenopathy are at increased risk of infradiaphragmatic disease.
Cervical adenopathy is less common in NHL, but commonly occurs in association with extranodal disease in Waldeyer’s ring. Approximately 40–60% of patients who present with head and neck involvement will have disseminated NHL. Involved nodal groups tend to be non-contiguous. Central necrosis within a lymph node is rarely seen. Imaging with contrast-enhanced CT or MRI has a useful role in evaluating the neck in patients with lymphoma, as it may identify enlarged nodes which are impalpable. It is helpful in response assessment, particularly in patients treated with radiotherapy, where post-treatment fibrosis renders clinical assessment difficult.
Thorax
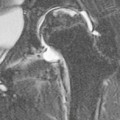
Intrathoracic nodes are involved at presentation in 60–85% of patients with HL and 25–40% of patients with NHL.19 Nodes larger than 1 cm short-axis diameter are considered enlarged. Any intrathoracic group of nodes may be affected, but all the mediastinal sites other than paracardiac and posterior mediastinal nodes are more frequently involved in HL than NHL. Nearly all patients with nodular sclerosing HL have disease in the anterior mediastinum. The frequency of nodal involvement in HL is as follows:19 prevascular and paratracheal—84% (Fig. 70-1); hilar—28% (Fig. 70-1); subcarinal—22% (Fig. 70-1); others—5% (aortopulmonary, anterior diaphragmatic, internal mammary (Fig. 70-2)). In NHL involvement of the hilar and subcarinal groups is rarer, occurring in 9 and 13%, respectively, whereas superior mediastinal nodes are involved in 35%.20


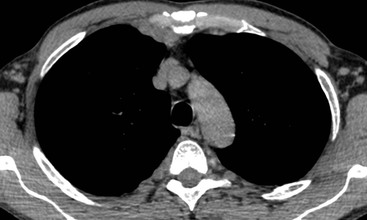
The great majority of cases of HL show enlargement of two or more nodal groups, whereas only one nodal group is involved in up to half of the cases of NHL. Hilar nodal enlargement is rare without associated mediastinal involvement, particularly in HL. Although paracardiac and internal mammary nodes are rarely involved at presentation in HL, they may be involved in recurrent disease, as they are not included in the classical ‘mantle’ radiation field. In HL and NHL, large anterior mediastinal masses usually represent thymic infiltration as well as a nodal mass (Fig. 70-3). A large anterior mediastinal mass in HL is recognised as an adverse prognostic feature and, as such, defines the need for more aggressive initial therapy. CT will demonstrate unsuspected mediastinal nodal enlargement despite a normal chest radiograph in 10% of patients with HL, and these patients have a poorer prognosis. CT of the chest has been shown to alter management in up to 25% of patients with HL. The therapeutic impact is less in patients with NHL, who are likely to receive chemotherapy regardless of stage. Impalpable axillary nodal enlargement is also frequently detected on CT in HL and NHL.


Abdomen and Pelvis
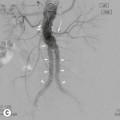
At presentation the retroperitoneal nodes are involved in 25–35% of patients with HL but up to 55% of patients with NHL.21 Mesenteric lymph nodes are involved in more than half the patients with NHL and less than 5% of patients with HL.21 Other sites such as the porta hepatis and splenic hilum are also less frequently involved in HL than NHL (Fig. 70-4). In HL, nodal spread is predictably from one lymph node group to another through directly connected lymphatic pathways. Nodes are frequently of normal size or only minimally enlarged. Spread from the mediastinum occurs through the lymphatic vessels to the retrocrural nodes, coeliac axis and so on. Around the coeliac axis, multiple normal-sized nodes may be seen, which can be difficult to evaluate because involved, normal-sized nodes are frequent in HL22 (Fig. 70-4). The coeliac axis, splenic hilar and porta hepatis nodes are involved in about 30% of patients and splenic hilar nodal involvement is almost always associated with diffuse splenic infiltration (Fig. 70-4). The node of the foramen of Winslow (porta caval node), lying between the portal vein and the inferior vena cava, is important, as it is often overlooked and may be the only site of disease relapse. It has a triangular shape; its normal long-axis diameter is up to 3 cm and in the anteroposterior plane is approximately 1 cm.

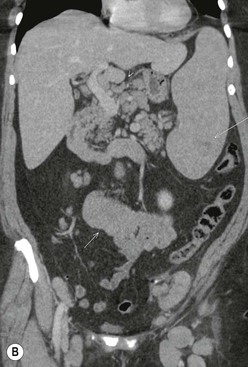
In NHL, nodal involvement is frequently non-contiguous and bulky and is more frequently associated with extranodal disease. Discrete mesenteric nodal enlargement or masses may be seen with or without retroperitoneal nodal enlargement. Large-volume nodal disease in both mesentery and retroperitoneum may give rise to the so-called ‘hamburger’ sign, in which a loop of bowel is compressed between two large nodal masses (Fig. 70-5). Multiple normal-sized mesenteric nodes should be regarded with suspicion for the diagnosis of lymphoma and lymphoma is a recognised cause of the ‘misty mesentery’. In NHL, regional nodal involvement is frequently seen in patients with primary extranodal lymphoma involving an abdominal viscus. Involved nodes tend to enhance uniformly and the presence of multilocular enhancement should suggest an alternative diagnosis such as tuberculosis or atypical infection.

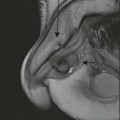
In the pelvis, any nodal group may be involved in both HL and NHL. Presentation with enlarged inguinal or femoral lymphadenopathy is seen in less than 20% of HL, and its presence should prompt close scrutiny of the pelvic nodal groups. In patients with massive pelvic disease, MRI is helpful for delineating the full extent of tumour and the effect on the adjacent organs.
Extranodal Disease in Lymphoma
Involvement of extranodal sites by lymphoma usually occurs in the presence of widespread advanced disease elsewhere. Such secondary involvement is a recognised adverse prognostic feature in HL and NHL but is much commoner in the latter. However, in approximately 35% of cases of NHL, primary involvement of an extranodal site occurs, with lymph node involvement limited to the regional lymph nodes: stages I–IIE. Primary extranodal HL is extremely rare and rigorous exclusion of disease elsewhere is essential before this diagnosis can be made. The incidence of extranodal involvement in NHL depends on factors such as the age of the patient, the presence of pre-existing immunodeficiency and the pathological subtype of lymphoma. Extranodal disease is commoner in children, (particularly in the gastrointestinal tract, the major abdominal viscera and extranodal locations in the head and neck)23 and in the immunocompromised host. The high incidence of extranodal involvement in these patient groups is a reflection of the fact that such lymphomas are usually aggressive histological subtypes.
The incidence of extranodal NHL is rising faster than that of nodal NHL. For example, primary lymphomas of the CNS were increasing in frequency at a rate of 10% per annum until the introduction of highly active antiretroviral therapies.24 Of the various pathological subtypes of NHL, mantle cell (a diffuse B-cell lymphoma), lymphoblastic lymphomas (80% of which are T-cell), BL (small cell non-cleaved) and MALT lymphomas demonstrate a propensity to arise in extranodal sites.
CT generally performs well in the depiction of extranodal disease, though there are certain instances where other techniques are preferable. FDG-PET is more sensitive than CT chiefly because of its ability to identify splenic and bone marrow infiltration13 (Fig. 70-6). PET or PET/CT can upstage as many as 40% of cases, though the CT component remains essential: for example, in low-grade lymphoma and in the lungs, where small nodules may be below the resolution of PET technology.


Thorax
Pulmonary Parenchymal Involvement
Several categories of lung involvement can be identified including:
• lymphomatous involvement associated with existing or previously treated intrathoracic nodal disease;
• lymphomatous involvement associated with widespread extrathoracic disease;
Some authors also separate out AIDS-related lymphoma (ARL) and the post-transplant lymphoproliferative disorder (PTLD),25 both of which commonly affect the lungs.
Lung involvement at presentation occurs in just under 4% of patients with NHL, but in approximately 12% of patients with HL. It is usually secondary to direct extension of nodal disease into the adjacent parenchyma, hence its paramediastinal or perihilar location. In this circumstance there is no effect on stage; the ‘E’ lesion. Patients with HL presenting with an intrapulmonary lesion in the absence of demonstrable mediastinal disease are unlikely to have lymphomatous disease of the lung unless there has been previous mediastinal or hilar irradiation, when recurrence may be confined to the lungs. Conversely, in NHL, nodal disease is absent in 50% of those patients with pulmonary or pleural involvement. As nodal disease progresses or relapses, lung involvement becomes commoner in HL and NHL, such that 30–40% of patients with HL have pulmonary involvement at some stage during the course of the disease.
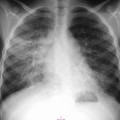
The radiographic appearances are extremely variable, but the commonest pattern is of one or more discrete nodules, with or without cavitation, which tend to be less well defined than those of primary or metastatic carcinoma, which they otherwise resemble (Figs. 70-7 and 70-8).26 The disease often spreads along lymphatic channels and involves lymphoid follicles around bronchovascular divisions, resulting in peribronchial nodulation spreading out from the hila, which can result in streaky shadowing visible on chest radiographs and at CT (Fig. 70-7).


Less commonly, lymphomatous cells fill the pulmonary acini, producing rounded or segmental areas of consolidation with air bronchograms (Fig. 70-8). Nodulation along the bronchial wall may enable differentiation from infective consolidation. A rare pattern of disease is widespread interstitial reticulonodular shadowing, producing a lymphangitic picture. Another rare manifestation is atelectasis, which usually results from endobronchial lymphoma rather than extrinsic compression by nodal disease.
The differential diagnosis of pulmonary involvement in lymphoma is extensive, and includes drug-induced changes, the effect of radiotherapy, and opportunistic infection during or following chemotherapy, particularly in patients with antecedent immunosuppression. Precise clinical correlation is essential in determining the most likely diagnosis.
Primary Pulmonary Lymphoma
Primary pulmonary lymphoma accounts for less than 1% of all lymphomas and is usually low-grade B-cell NHL, arising from MALT or bronchus-associated lymphoid tissue (BALT). BALT lymphomas tend to occur in the fifth to sixth decades, have an indolent course with 5-year survivals of over 60% and tend to remain extranodal, although lymph node involvement can occur with advanced disease.27 Many patients will have a prior history of inflammatory or autoimmune disease, such as Sjögren’s syndrome, collagen vascular disease and dysgammaglobulinaemia.28 The imaging findings are non-specific with the single commonest manifestation being a solitary nodule. Multiple nodules, or one or more rounded or segmental areas of consolidation,29 are also seen. These can persist unchanged for long periods. Pleural effusions are seen in up to 20% of cases.28
In the remaining 15–20% of patients, primary lung lymphoma is due to high-grade NHL. The most common finding on a chest radiograph is of a solitary or multiple pulmonary nodules, which characteristically grow rapidly. Chest wall and nodal involvement occurs more frequently than with pulmonary MALT-type lymphomas.27 Primary pulmonary HL is extremely rare. The most frequently described finding is single or multiple nodules with upper zone predominance and a relatively high incidence of cavitation.
Pleural Disease
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


