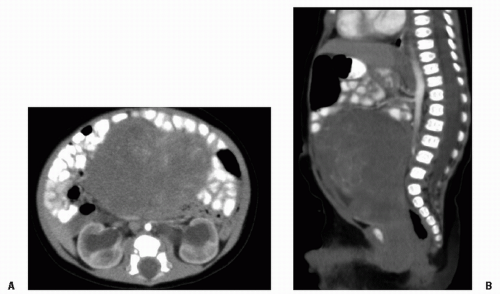
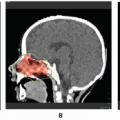
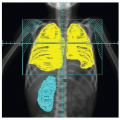
Rhabdomyosarcoma (RMS) is a highly malignant neoplasm that arises from embryonal mesenchyme with the potential for differentiating into striated muscle. Although the cells show differentiation along rhabdomyoblastic lines, RMS is not limited to cells with recognizable muscle cross-striations (
1,
2). Weber initially described RMS in 1854, but progress in our understanding of this complex neoplasm accelerated with Stout’s landmark descriptive series in 1946 and the delineation by Horn and Enterline in 1958 of the four classic forms of RMS (
3,
4,
5). RMS can arise almost anywhere in the body, is locally invasive, and rapidly disseminates early in its course. Near the beginning of the 20th century, the only cures were accomplished with radical surgery in the few fortunate children without metastases. Significant disfigurement and loss of function were common sequelae. High-dose radiation therapy (RT) increased the potential for local control but caused a different set of morbidities (
6,
7). As chemotherapy has become increasingly effective in eliminating micrometastatic disease and assisting in local control, the need for aggressive surgery and large-volume irradiation has diminished, although surgery and RT still play pivotal roles in the curative treatment of RMS (
8). Overall survival rates have concomitantly increased from 15-25% to more than 70% (
9,
10,
11,
12).
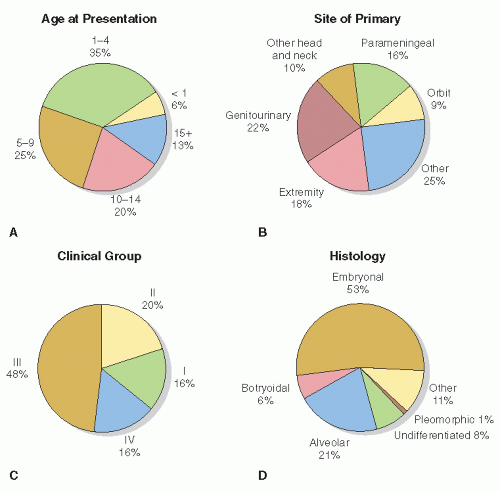
RMS is a rare tumor with clinical and biologic heterogeneity. Consequently, multi-institutional trials were necessary to develop and refine treatment approaches. North American investigators of the Intergroup Rhabdomyosarcoma Study Group (IRSG) have played a paramount role in this progress, now in its sixth generation of protocols. The difficulty of this undertaking is clear in view of the myriad sites, stages, and histologies of RMS that are associated with different natural histories and prognoses (
13). Beyond this, advances in imaging, changes in endpoints, and the need for mature data all increased the difficulty in conducting and comparing randomized, controlled clinical studies of patients with RMS. With this view, the IRSG was established through the collaboration of three multidisciplinary cancer treatment study groups (Cancer and Leukemia Group B, Children’s Cancer Study Group, and the Pediatric Branch of the Southwest Oncology Group, which later became the Pediatric Oncology Group). Intergroup Rhabdomyosarcoma Study I (IRS-I) was open for patient entry from 1972 to 1978 (
9). With an overall 5-year survival rate of 55% in IRS-I, IRS-II was designed to improve survival for the patient subgroups with poor outcomes and to refine treatment for the remaining patients (
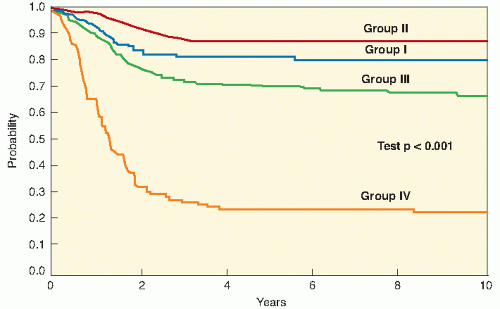
10). IRS-II ran from 1978 to 1984, IRS-III from 1984 to 1991, IRS-IV from 1991 to 1997, and IRS-V from 1997 to 2005-2006. Although the previous generations of IRS studies were based on a surgically oriented clinical grouping system dependent on the tumor that remained after initial surgery, IRS-IV and IRS-V were based on a more biologically oriented staging system, discussed later in this chapter. These studies, each based on the results of its predecessor, have provided a database of over 4000 patients. The IRSG studies are now conducted through the Children’s Oncology Group, the United States-based cooperative research network that formed from the merger of the Pediatric Oncology Group and the Children’s Cancer Study Group. The current generation of studies opened in 2005-2006, upon the completion of IRS-V. Other multi-institutional group studies have also provided important data (
14,
15,
16,
17). Of particular note are the Europeanbased International Society of Pediatric Oncology (SIOP) and the United Kingdom-based Children’s Solid Tumor Group (CSTG) (
15,
16,
17). Finally, many single-institution studies have also been quite informative regarding RMS (
7,
8,
18,
19). Progress in treating RMS is exemplified by the improvement in overall 5-year survival in the IRS studies: 55% in IRS-I, 63% in IRS-II, and 71% in IRS-III (
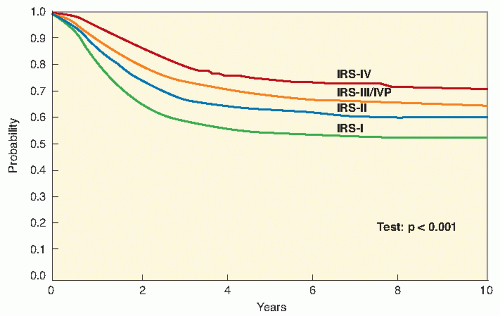
Fig. 11.1) (
9,
10,
11,
20). Early results from IRS-IV show an overall 3-year survival rate of 86% for patients with nonmetastatic disease and 39% for patients with metastatic disease (
12,
21).