TABLE 5.1 Small Round Cell Tumors of Bone | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||
Monostotic or polyostotic histiocytic and dendritic monoclonal cell disorder characterized by neoplastic proliferation of Langerhans cells that normally populate the skin, mucosal surfaces, lymph nodes, and other tissues
Relatively rare disorder, accounting for less than 1% of all osseous lesions.
Age distribution ranging from the first month of life to eighth decade, with 80% to 85% of cases seen in patients under the age of 30, and 60% under the age of 10.
Males are affected twice as often as females.
Any bone may be affected, although there is predilection to involve the bones of the skull, particularly the calvaria.
Other commonly involved sites include the femur, the bones of the pelvis, ribs, vertebrae, and the mandible.
Pain and swelling of the affected area occur most commonly.
In cases of temporal bone involvement, the presenting features can overlap with those of otitis media and mastoiditis.
Mandibular involvement may result in loosening and loss of teeth.
Involvement of vertebral body may result in compression fracture and possible neurologic impairment.
Fever, elevated sedimentation rate, and leukocytosis may be present.
TABLE 5.2 Small Round Cell Tumors of Bone and Typical Immunohistochemistry Results
Tumor
bcl-2
CK
Vim
Des
NF
CD99
CD57
NSE
CD45
Light Chains κ or λ
Syn
CD138
Ewing sarcoma
–
+/a
+
–
-/a
+
(+)
–
–
–
–
–
PNET/Askin tumor
–
–
+
–
(+)
+
(+)
+
–
–
+
–
Malignant lymphoma
+
–
+
–
–
(+)
(+)
–
+
+/-
–
(+)
Myeloma
+
–
-/+
–
–
(+)
–
–
-/+
+
–
+
Small cell osteosarcoma
–
–
+
–
–
–
–
–
–
–
–
–
Mesenchymal chondrosarcoma
–
–
+
–
–
+
–
–
–
–
–
–
Metastatic neuroblastoma
–
–
–
–
+
–
(+)
+
–
–
+
–
Rhabdomyosarcoma
–
–
(+)
+
–
–
–
(+)
–
–
–
–
Synovial sarcoma
+
+
+
–
–
+
–
–
–
–
–
(+)
Desmoplastic small round cell tumor
–
+
+
+
–
(+)
+
+
–
–
+
–
From Greenspan A, Jundt G, Remagen W. Differential Diagnosis in Orthopaedic Oncology. 2nd ed. p. 333 , Table 5-2.
TABLE 5.3 Revised European-American Lymphoma Classification
B-Cell Lymphomas
T-Cell and Natural Killer Cell Neoplasms
Hodgkin Disease
Precursor B-cell neoplasm
▪ Precursor B-lymphoblastic leukemia or lymphoma
Mature B-cell neoplasm
▪ B-cell chronic lymphocytic leukemia, prolymphocytic leukemia, small lymphocytic leukemia
▪ Lymphoplasmacytoid lymphoma
▪ Mantle cell lymphoma
▪ Follicle center lymphoma
▪ Marginal zone B-cell lymphoma
▪ Hairy cell lymphoma
▪ Diffuse large cell B-cell lymphoma
▪ Burkitt lymphoma
▪ High-grade B-cell lymphoma
Precursor T-cell neoplasm
▪ Precursor T-lymphoblastic lymphoma or leukemia
Peripheral T-cell and natural killer cell neoplasm
▪ T-cell chronic lymphocytic leukemia
▪ Large granular lymphocyte leukemia
▪ Mycosis fungoides, Sézary syndrome
▪ Peripheral T-cell lymphoma
▪ Angioimmunoblastic T-cell lymphoma
▪ Angiocentric lymphoma
▪ Adult T-cell lymphoma
▪ Anaplastic large cell lymphoma
Nodular lymphocyte predominance (paragranuloma)
Nodular sclerosis
Mixed cellularity
Lymphocyte depletion
Lymphocyte-rich classic Hodgkin disease
From Greenspan A, Jundt G, Remagen W. Differential Diagnosis in Orthopaedic Oncology. 2nd ed. p. 335 , Table 5-3. Modified from Krishnan A, Shirkhoda A, Tehranzadeh J, et al. Primary bone lymphoma: radiographic—MR imaging correlation. Radiographics. 2003;23:1371-1387, with permission.
Single or multiple lesions restricted to the skeleton have been termed eosinophilic granuloma.
Multifocal bone disease associated with exophthalmos and diabetes insipidus is known as Hand-Schuller-Christian disease.
Letterer-Siwe disease (nonlipid reticulosis) is a condition that usually affects very young children (<2 years old) and consists of disseminated bone lesions, anemia, lymphadenopathy, and splenomegaly.
Radiography shows well-defined, lytic lesions; however, in a minority of cases, the lesions may exhibit wide zone of transition and permeative type of bone destruction (Figs. 5.1, 5.2 and 5.3). More sclerotic appearance is seen in later stages of the disease (Fig. 5.4). Some lesions may show slanting or beveling of the edges. Scalloping of the endocortex is a common finding (see Fig. 5.1B).
Cortical involvement may elicit an aggressive lamellated (onion-skin) type of periosteal reaction (Fig. 5.5A, see also Fig. 5.1A).
Scintigraphy generally shows increased uptake of the radiopharmaceutical tracer (see Fig. 5.6B), but about 35% of lesions show normal radionuclide bone scan.
Involved tissue is soft and red in color.
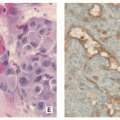
Proliferating Langerhans cells are arranged in aggregates, sheets, or individually within a loose fibrous stroma, exhibiting indistinct cytoplasmic borders and eosinophilic to clear cytoplasm (Fig. 5.9).
The nuclei are translucent, ovoid, coffee bean or kidney shaped with typical longitudinal grooves (Fig. 5.9C).
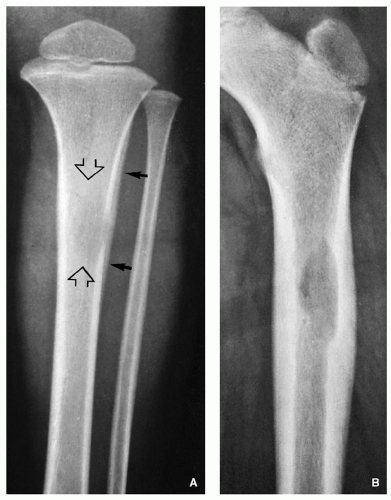
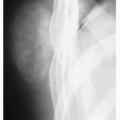
FIGURE 5.1 Radiography of Langerhans cell histiocytosis. (A) Anteroposterior radiograph of the lower leg of a 4-year-old boy shows a lesion in the diaphysis of the left tibia (open arrows). Observe a permeative type of bone destruction and a lamellated (onionskin) type of periosteal reaction (arrows), very similar to that seen in Ewing sarcoma. (B) Anteroposterior radiograph of the proximal right femur of a 3-year-old boy shows an osteolytic lesion causing endosteal scalloping. Note fusiform thickening of the cortex and solid uninterrupted periosteal reaction.
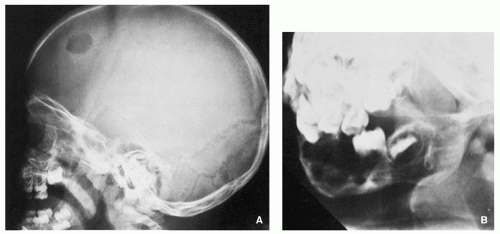
FIGURE 5.2 Radiography of Langerhans cell histiocytosis. (A) Lateral radiograph of the skull of a 2.5-year-old boy with disseminated disease shows an osteolytic lesion in the frontal bone with a sharply outlined borders, exhibiting a “punched-out” appearance. Uneven involvement of the inner and outer tables results in beveled presentation. (B) A 3-year-old girl with extensive skeletal involvement had a large destructive lesion in the mandible. Note the characteristic appearance of a floating tooth, which resulted from destruction of supportive alveolar bone.
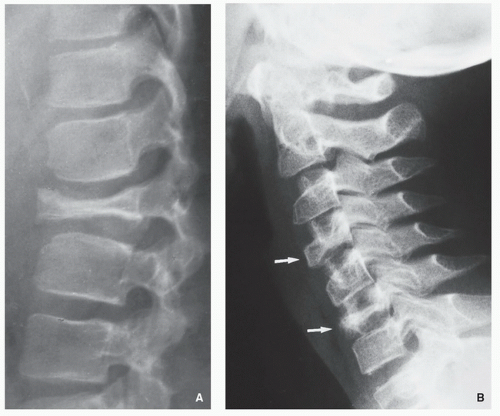
FIGURE 5.3 Radiography of Langerhans cell histiocytosis. (A) Vertebra plana represents collapse of vertebral body secondary to destruction of bone by granulomatous lesion. Note the preservation of intervertebral disk space. (B) In another patient, lateral radiograph of the cervical spine shows compression fractures of the vertebral bodies C4 and C6 (arrows).
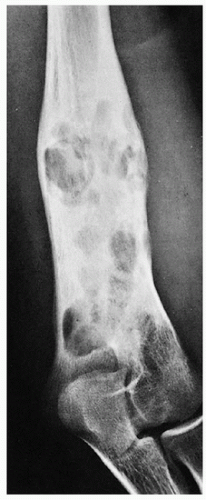
FIGURE 5.4 Radiography of Langerhans cell histiocytosis. The healing stage of the lesion, as seen here in the distal humerus of a 16-year-old girl, exhibits predominantly sclerotic changes with interspersed radiolucent foci, thickening of the cortex, and well-organized periosteal reaction. In this stage, the lesion mimics chronic osteomyelitis.
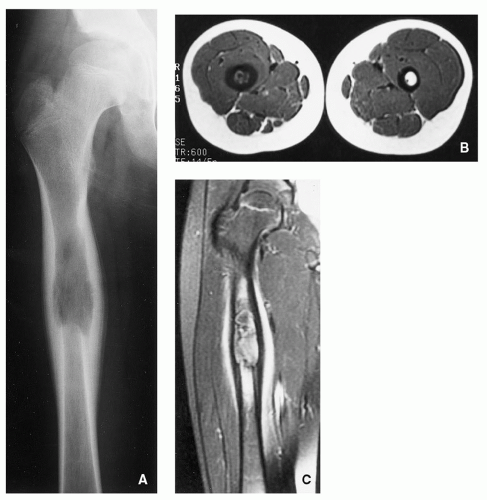
FIGURE 5.5 Radiography and magnetic resonance imaging of Langerhans cell histiocytosis. (A) Anteroposterior radiograph of the right femur of a 13-year-old boy demonstrates a radiolucent lesion in the proximal femoral diaphysis with a lamellated periosteal reaction. (B) Axial T1-weighted MRI shows the lesion to be of low signal intensity. Observe marked thickening of the cortex. (C) Coronal T1-weighted MR image obtained after intravenous administration of gadolinium shows marked enhancement of the lesion and soft tissues adjacent to the thickened cortex.
Chromatin is either diffusely dispersed or condensed along the nuclear membranes.
Langerhans cells are frequently admixed with inflammatory cells including large numbers of eosinophils, lymphocytes, neutrophils, and plasma cells (Figs. 5.10 and 5.11).
Necrosis may be found in a minority of cases, and if it is prominent, is usually the complication of a pathologic fracture.
Mitotic figures may be seen; however, atypical forms are absent.
In older or polyostotic lesions, lipid-bearing foam cells can be observed (see Fig. 5.9D).
Special stains may reveal abundant droplets of sudanophilic fat peripherally or in the middle of the giant cell cytoplasm, so-called Touton cells.
Langerhans cells are positive for CD1a, S-100 protein (see Figs. 5.10C and 5.11C,D), and Langerin/CD207, but negative for CD68 and CD45.
Intracytoplasmic “tennis racquet”-shaped inclusion bodies (organelles), known as Birbeck granules, are diagnostic for this disorder (Fig. 5.12).
Treatment and prognosis of LCH depends on the site and size of the lesion, the age of the patient, and the presence or absence of multifocal disease.
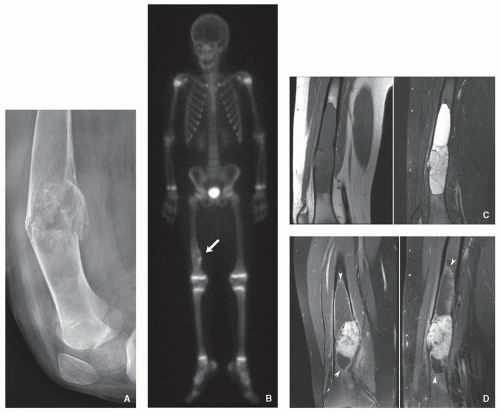
FIGURE 5.6 Radiography, scintigraphy, and magnetic resonance imaging of Langerhans cell histiocytosis. (A) Lateral radiograph of the right knee of a 9-year-old boy shows an osteolytic lesion in the diaphysis of the distal femur with a pathologic fracture. (B) Whole-body radionuclide bone scan obtained after intravenous injection of 15 mCi of 99mTc-labeled methylene diphosphonate shows mildly increased uptake of the radiopharmaceutical agent at the site of the lesion (arrow). There are no additional lesions present. (C) Sagittal T1-weighted and sagittal STIR MR images demonstrate sharply demarcated lesion exhibiting low signal intensity on T1 weighting and high signal intensity on STIR sequence. (D) Coronal and sagittal T1-weighted fat-suppressed MR images obtained after intravenous administration of gadolinium show significant enhancement of the lesion. The solid, enhancing lesion is surrounded proximally and distally by intramedullary cyst formation with a thin enhancing peripheral rim (arrowheads), a rare feature of this disorder.
Monostotic disease is usually managed by curettage; however, tumors located in areas difficult to excise may be treated with low dose of radiation.
Single- or multi-agent therapy may be administered in the setting of disseminated disease.
Complete resolution may follow treatment or occasionally may occur spontaneously.
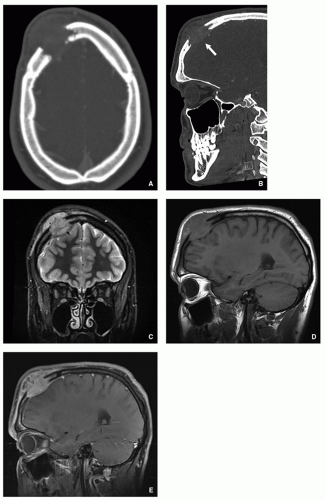 FIGURE 5.7 Computed tomography and magnetic resonance imaging of Langerhans cell histiocytosis. Axial (A) and sagittal (B) reformatted CT images of the skull of a 19-year-old man show a destructive lesion in the right frontal bone associated with a soft-tissue mass (arrow). (C) Coronal T2-weighted fatsuppressed MR image shows a soft-tissue mass to better advantage, displaying high signal intensity. The mass is compressing the right frontal lobe. (D) Sagittal T1-weighted and (E) sagittal T1-weighted fat-suppressed MRI obtained after intravenous administration of gadolinium show slight enhancement of both the intraosseous lesion and the softtissue mass. |
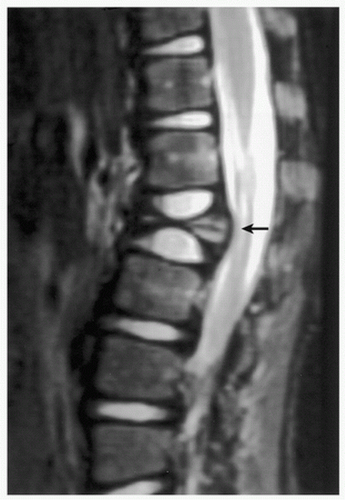 FIGURE 5.8 Magnetic resonance imaging of Langerhans cell histiocytosis. A sagittal T2-weighted MR image shows compression of the ventral aspect of the thecal sac by fractured vertebral body (arrow). |
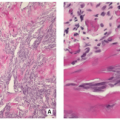
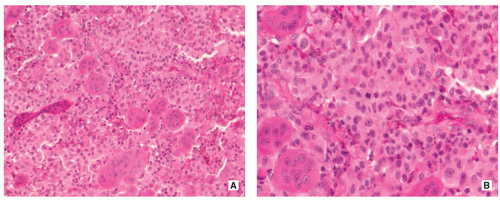 FIGURE 5.9 Histopathology of Langerhans cell histiocytosis. (A) Densely arranged large Langerhans cells and giant cells are infiltrated by numerous eosinophilic granulocytes (H&E, original magnification ×100). (B) At higher magnification large pale histiocyte-like Langerhans cells and numerous giant cells containing up to 10 nuclei (left) are clearly discernible. In addition, sparse lymphocytes and eosinophils are present (H&E, original magnification ×400). |
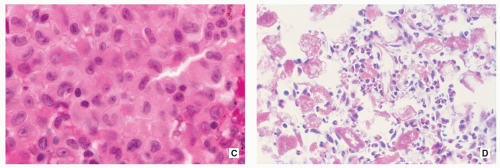 FIGURE 5.9 (Continued). (C) At high power, the indentations of oval nuclei of Langerhans cell are clearly visible (H&E, original magnification ×630). (D) In another field of view, mulberry-like foam cells (left and upper right), eosinophilic granulocytes, and hyaline bodies prevail (H&E, original magnification ×50). |
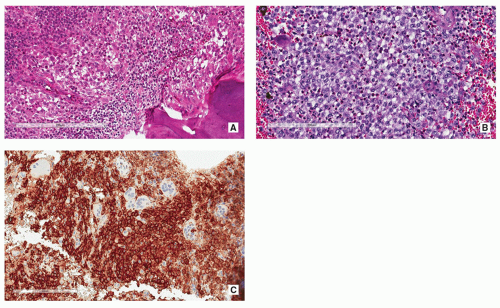 FIGURE 5.10 Histopathology of Langerhans cell histiocytosis. (A) Aggregates of ovoid to round histiocyte-like Langerhans cells are present. Note also prominent eosinophilia and foci of hemorrhage (H&E, original magnification ×100). (B) Higher magnification shows Langerhans cells in a mixed inflammatory background of lymphocytes, plasma cells, and eosinophils (H&E, original magnification ×200). (C) Immunohistochemistrγ shows membrane-based positivity of Langerhans cells for CD1a (original magnification ×200). |
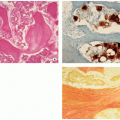
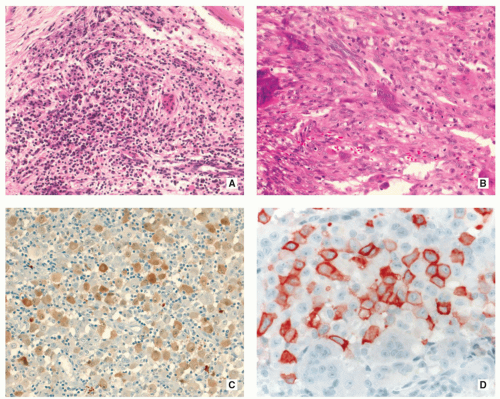 FIGURE 5.11 Histopathology of Langerhans cell histiocytosis. (A) In a later stage of development, nonspecific inflammatory cells, particularly lymphocytes, predominate. Some giant cells may also be present (H&E, original magnification ×100). (B) In another field, Langerhans cells show a more spindle-shaped appearance. Scattered eosinophils and giant cells are also present (H&E, original magnification ×200). (C) Both cytoplasm and nuclei of the tumor cells are positive for S-100 protein (biotin-streptavidin peroxidase, original magnification ×200). (D) On higher magnification observe cytoplasmic and membranous positivity of Langerhans cells for CD1a (ABC peroxidase, original magnification ×400). |
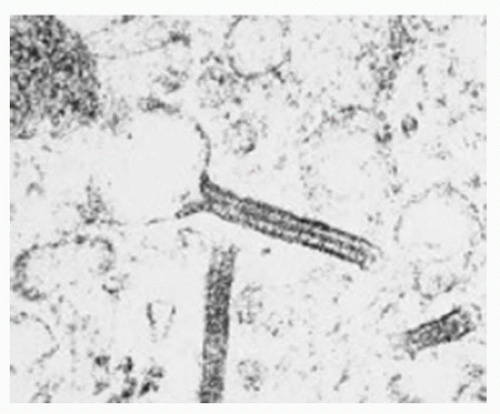 FIGURE 5.12 Electron microscopy of Langerhans cell histiocytosis. Intracytoplasmic tennis racquet-shaped inclusions known as Birbeck granules are pathognomonic for this disorder (EM, original magnification ×64,000). |
Also known as sinus histiocytosis with massive lymphadenopathy, this rare, proliferative, histiocytic disease of unknown etiology is characterized by the enlargement of lymph node sinuses caused by an aggregation of histiocytic cells that exhibit marked lymphophagocytosis (numerous phagocytized lymphocytes are present in the cytoplasm).
Majority of patients are teenagers and young adults.
Mean age of presentation is 20 years.
No gender preference.
Solitary lesion in bones was described in young children.
Tibia, femur, clavicle, skull, maxilla, calcaneus, metacarpals, and sacrum.
Fever and massive cervical lymphadenopathy are the most common symptoms at presentation.
Other symptoms include weight loss, malaise, and night sweats.
Quite often the disease fully manifests after a short period of a nonspecific fever and pharyngitis.
Primary or secondary involvement of extranodal sites, including the skeleton, is common.
Skeletal involvement manifests by the presence of solitary or multifocal defects with poorly or well-demarcated (sclerotic) borders.
The intramedullary lesions are associated with cortical erosion, complete cortical disruption, periosteal reaction, or a combination of these features.
Radiographic manifestations and clinical symptoms may suggest an inflammatory disorder, such as osteomyelitis.
Scintigraphy using gallium scanning shows increased uptake of the radiopharmaceutical agent.
FDG-PET scanning shows increased metabolism.
In typical cases, the sinuses of lymph nodes are filled with large, pale histiocytic cells of varying size.
These cells have prominent eosinophilic cytoplasm, indistinct borders, and round or oval nuclei with a very fine chromatin pattern and a single small nucleolus.
Nuclear grooves are not present, and some of these cells may have several nucleoli.
Occasionally, cells with multilobulated nuclei may be present.
Mitotic figures are rare; atypical mitoses are not present.
The most striking and diagnostically important feature of histiocytic cells is prominent emperipolesis or lymphophagocytosis (i.e., the presence of well-preserved lymphocytes within the histiocytic cell cytoplasm) (Fig. 5.13A).
In addition to lymphocytes, a smaller number of phagocytized plasma cells, neutrophils, and red cells are also present.
Extranodal disease, including skeletal system, has all these features except that histiocytic cells, instead of growing in sinuses, form irregular geographic areas separated by other inflammatory cells.
Histiocytic cells are positive for S-100 protein (Fig. 5.13B) but negative for CD1a.
This disease is considered a histologically benign, proliferative, histiocytic disorder with a variable, but occasionally fatal, outcome.
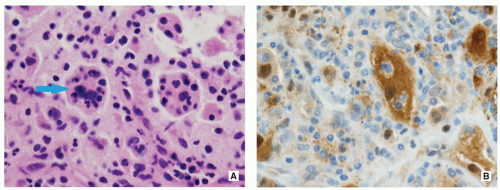
FIGURE 5.13 Histopathology of Rosai-Dorfman disease. (A) Large histiocytic cell with well-preserved lymphocytes within its cytoplasm (blue arrow), also known as emperipolesis or lymphophagocytosis, is a pathognomonic feature of this disorder (H&E, original magnification ×400). (B) Immunohistochemistry demonstrates strong positivity of large histiocytes to S-100 protein (original magnification ×400).
The majority of patients have indolent regressive or clinically stable disease after several years.
Fatal outcome of the disease is associated with the severe involvement of extranodal sites (lungs and kidney).
Rare disseminated histiocytic disorder of unknown cause characterized by infiltration of musculoskeletal system and various organs including the heart, lungs, and skin by lipidladen histiocytes leading to fibrosis and osteosclerosis.
Slight male predominance.
Age range between 7 and 84 years with mean age of 53.
Mostly affects the major long bones with sparing of the articular ends.
Rarely the flat bones can be involved.
Extraskeletal involvement may occur, for example, kidney, heart, lungs, and skin.
Mild bone pain sometimes associated with soft-tissue swelling.
Extraskeletal manifestations may include general weakness, fever, weight loss, abdominal pain, shortness of breath, neurologic dysfunction, exophthalmos, diabetes insipidus, kidney failure, hepatosplenomegaly, and eyelid xanthomas.
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


