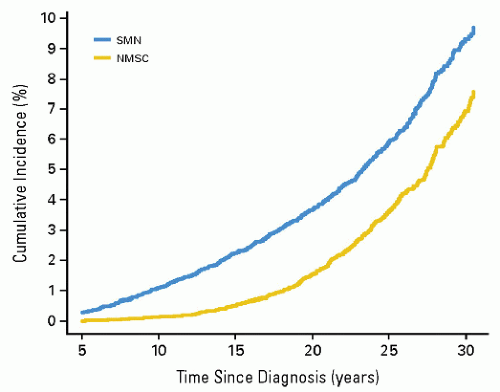
 Figure 20.1 Cumulative incidence of second malignant neoplasms and nonmelanoma skin cancers in childhood cancer survivors. (From Meadows AT, Friedman DL, Neglia JP, et al. Second neoplasms in survivors of childhood cancer: findings from the Childhood Cancer Survivor Study cohort. J Clin Oncol. 2009;27:2356-2362, with permission). |
treatment. Follow-up of the Childhood Cancer Survivor Study demonstrates that, after adjusting for exposure to radiation, the risk of SPC was significantly associated with female sex (p < 0.001), childhood cancer diagnosed at a younger age (p < 0.001), initial cancer diagnosis of Hodgkin lymphoma (p < 0.001), and exposure to alkylating agents (p = 0.02) (6).
Table 20.1 Second Cancers and their Relationship with Primary Cancers | ||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
A variety of histologic types of neoplasms can be induced by irradiation. These cancers are indistinguishable morphologically from naturally occurring cancers. The identification of a “radiation signature” in tumors would be important in evaluating SPC. There is evidence that radiation produces a different spectrum of mutations than other genotoxic agents. If these mutations could be discerned and characterized, then one could clearly identify radiation-induced tumors and understand, more completely, the radiation dose-response relationship for induced tumors. In this manner, molecular forensics may affect our understanding of attributable risk (25). Of interest is recent data showing that the risk of an SPC is proportional to the number of premalignant stem cells created, and surviving RT. This is related to cell killing by RT, cellular repopulation between RT fractions and after the last fraction, and the ratio of the proliferation rate of premalignant cells to the proliferation rate of normal cells (Fig. 20.2).
Low-linear energy transfer (low-LET) radiation (gamma ray, x-ray) is generally less efficient in inducing tumors than high-LET radiation. In a murine hepatocarcinogenesis model, for example, neutron irradiation produced a greater incidence of hepatomas than did gamma irradiation (26). Low-LET radiation appears to become less effective at carcinogenesis per centigray as the dosage falls, whereas high-LET radiation (neutrons, alpha particles) does not (27). With low-LET irradiation there is less tumor induction when the dosage is fractionated or administered at low dose rates; implying that repair of carcinogenic damage is occurring. With high-LET irradiation, the radiobiological effect is higher at low dosages. The carcinogenic effectiveness of high-LET radiation is not diminished and may be increased by dosage fractionation or protraction (28).
Comparison of the frequency of SPCs in different centers suggests that orthovoltage therapy is more likely to be carcinogenic than megavoltage therapy (24,29). This may be dose-related: By delivering a higher dosage to bone, orthovoltage irradiation may increase the risk of an SPC of bone. The higher risk may also be related to the long follow-up available for orthovoltage patients.
Although every tissue in the body is at risk for radiation-induced cancer, sensitivity varies according to the tissue. For example, thyroid gland and breast are sensitive to cancer induction after low radiation dosages; lymphoid tissue, lung, and liver are susceptible at moderate dosages, and bone at higher dosages. The relationship between dosage and response may vary according to the induced tumor. For example, the development of CNS tumors increases with increasing dose, whereas secondary thyroid carcinomas have a peak incidence after doses of 20-29 Gy but then decrease (Fig. 20.3A,B). Cancer risk from radiation may be given per unit dosage (gray, Gy) or per unit dosage equivalent (Sievert, Sv) where a “quality factor” (Q) is used to take account of the varying biological effectiveness of different radiations (e.g., for a gamma ray Q = 1, and for a neutron Q = 20). Therefore, dosage in Sv = dosage in Gy × Q. Detailed literature reviews indicate that the cancer mortality risk for the general population after whole body exposure is 1 × 10−4 to 4 × 10-4 per person-cGy. Pooled results of various partial body exposures give an estimated risk of 1 × 10-4 to 4 × 10-4 per person-cGy (4 × 102 Sv1). The risk based on incidence is about twice that for mortality (27,30). Leukemia data can be fit by a curvilinear dose-response model, whereas skin cancer appears to have a threshold dose-response function, and breast data fit a linear no-threshold model.

Figure 20.2 Initiation/inactivation/proliferation model of carcinogenesis. Risk of SMN proportional to the number of premalignant stem cells created and surviving RT. Related to: (1) cellular killing, (2) cellular repopulation occurring between fractions and after the last fraction, and (3) the ratio of proliferation rate for premalignant cells to the proliferation rate of normal cells. (From Sachs RK, Brenner DJ. Solid tumor risks after high doses of ionizing radiation. Proc Natl Acad Sci U S A. 2005;102:13040-13045, with permission).
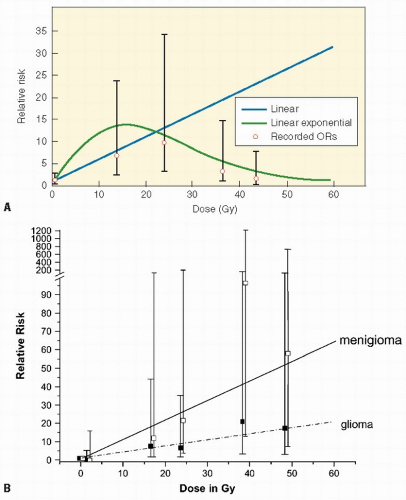
Figure 20.3 A: Dose-response relations between radiotherapy dose and relative risk of second neoplasms. (From Meadows AT, Friedman DL, Neglia JP, et al. Second neoplasms in survivors of childhood cancer: findings from the Childhood Cancer Survivor Study cohort. J Clin Oncol. 2009;27: 2356-2362) B: Dose-risk for RT-induced CNS tumors from Childhood Cancer Survivor Study of 14,361 5-year survivors. (From Neglia JP, Robison LL, Stovall M, et al. New primary neoplasms of the central nervous system in survivors of childhood cancer: a report from the Childhood Cancer Survivor Study. J Natl Cancer Inst. 2006;98:1528-1537, with permission).
Radiation induces solid SMNs, within the radiation field and in a dose-dependent fashion (6,11,16,31, 32, 33, 34). A recent report assessed the relationship between the radiation dose received inside of the edges of radiation beams throughout the volume of tissue irradiated (thus the integral dose) and the likelihood of developing a secondary malignancy. In a cohort of 4401 patients who were 3-year survivors of all types of childhood cancer, a dose-response relationship was found between the overall risk of an SMN and the estimated integral dose (Table 20.2, Fig. 20.4) (35). As a corollary, data from Diallo et al. showed that a greater volume of tissues receives low or intermediate radiation doses in regions bordering the irradiated volume with modern multiplebeam radiation therapy administrations, including intensity modulated radiation therapy (IMRT) (36). However, in a report that modeled risk for children treated with IMRT versus conventional therapy, an increase was not seen (37). Additional data will be necessary to settle this issue.
SPCs may also be induced by agents other than radiation (chemotherapy, environmental exposures, hereditary disposition). The low-dose radiation dose-response curve could be influenced by these confounding factors to produce a result that is the sum of two independent rates or may be greater than simple addition would indicate. A variety of chemotherapeutic agents (to be discussed), especially alkylators, are known to be carcinogenic, and they may be additive or synergistic with radiation. Immunosuppressive agents clearly influence the propensity for tumor induction, as is seen in the setting of organ transplantation (38).
A large number of irradiated patients are necessary to calculate a radiation carcinogenesis risk with reasonable accuracy.
Latent periods vary according to the induced tumor. At least two patterns of latent periods for radiation-induced cancer have been described. The first, exemplified by the risk of leukemia in atomic bomb survivors, consists of an early wavelike pulse of increased risk followed by a gradual decline to baseline levels. The second, more typical of solid tumors, is an increase in relative risk of SPCs over many years, which remains constant over time thereafter. The latter pattern suggests that a multi-event pattern of carcinogenesis is involved: the initial event (radiation) is followed by promoting events (i.e., smoking, alcohol, or environmental exposures) over many years (39). Long latent periods complicate the study of radiation-induced cancer because the presence of other carcinogens or disease processes may not be well documented.
The duration of follow-up for any study population influences the frequency of tumors seen.
It is unclear whether the risk of tumor development after exposure is simply an absolute increase that is proportional
to the radiation dosage or a relative increase that builds on an underlying spontaneous risk (greater for some patients) of developing a malignancy.
Age is a critical factor in determining radiation risk. In children, the most common SPCs occur in tissues undergoing rapid proliferation such as bone, thyroid, and breast. An actively proliferating tissue may be more susceptible to malignant transformation in any single cell because of the greater number of cell divisions. This might explain the higher frequency of secondary bone tumors in children than in adults (40). Childhood cancer survivors are likely to develop leukemias or sarcomas, whereas secondarily induced embryonal tumors are rare.
Table 20.2 Integral Dose as a Risk for Second Cancers | ||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||
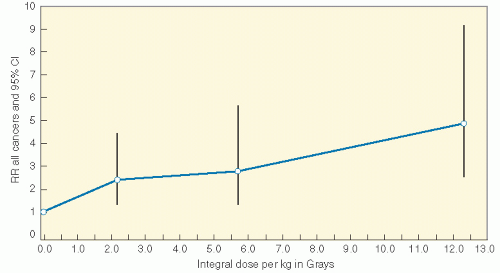 Figure 20.4 Integral dose and SMN risk in pediatric cancer survivors. (Modified from Nguyen F, Rubino C, Guerin S, et al. Risk of a second malignant neoplasm after cancer in childhood treated with radiotherapy: correlation with the integral dose restricted to the irradiated fields. Int J Radiat Oncol Biol Phys. 2008;70:908-915, with permission). |
ifosfamide, mechlorethamine, melphalan, busulfan, nitrosoureas, chlorambucil, dacarbazine (60), and platinum compounds (61). Mutagenicity is related to the ability of alkylating agents to form crosslinks and/or transfer alkyl groups to form DNA monoadducts. Alkylation results in inaccurate base pairing during replication and single- and double-strand breaks in the double helix as the alkylated bases are repaired. The risk of alkylating agent-related t-MDS/AML is dose-dependent, with a latency of 3-5 years after exposure (62); it is associated with abnormalities involving chromosomes 5 (− 5/del[5q]) and 7 (−7/del [7q]), and a high frequency of multidrug-resistance phenotype (63). The 5q31-33 region of the long arm of chromosome 5 contains at least nine genes involved in hematopoiesis. Defects in any of these genes could disrupt the balance between cell growth and differentiation and play a role in initiation and progression of leukemia. Complete or partial deletions of the long arm of chromosome 7 (7q— and -7) are nonrandom abnormalities observed in therapy-related leukemia.
Table 20.3 Characteristics of Therapy-Related Myelodysplasia or Acute Myeloid Leukemia After Treatment with Alkylating Agents and Topoisomerase II Inhibitors | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
2.2% at the low (less than 1.5 g/m2), moderate (1.5-3.0 g/m2), and high (more than 3.0 g/m2) dosages of etoposide, respectively, again suggesting a lack of dose-response relationship. A recent study using a case-controlled design explored the dose-response relationship between epipodophyllotoxins and anthracyclines, and the risk of developing secondary leukemia. This study concluded that patients who received 1.2-6.0 g/m2 of epipodophyllotoxins or more than 170 mg/m2 of anthracyclines have a risk seven times higher than that of patients who received lower dosages or none of these drugs (72).
Table 20.4 Features of Chemotherapy-Induced Hematopoietic Malignancies | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
RAD51-G-135C polymorphism is significantly over-represented in patients with t-MDS/AML compared with controls (C allele: OR = 2.7) (94). Although XRCC3-Thr241Met was not associated with t-MDS/AML (OR = 1.4, 95%CI, 0.7-2.9), a synergistic effect resulting in an eightfold increased risk of t-MDS/AML (OR = 8.1, 95% CI, 2.2-29.7) was observed in the presence of XRCC3-241Met and RAD51-135C allele in patients with t-MDS/AML compared with controls (94). Base excision repair (BER) pathway corrects individually damaged bases occurring as a result of ionizing radiation and exogenous xenobiotic exposure. The XRCC1 protein plays a central role in the BER pathway and also in the repair of single strand breaks, by acting as a scaffold and recruiting other DNA repair proteins (86,95). The protein also has a BRCA1 C-terminus (BRCT) domain—a characteristic of proteins involved in DNA damage recognition and response. The presence of variant XRCC1-399Gln has been shown to be protective for t-MDS/AML (96) and basal cell carcinoma (97). Nucleotide excision repair (NER) removes structurally unrelated bulky damage induced by radiation and chemotherapy. The polymorphic Gln variant (ERCC2 Lys751Gln) of the NER pathway is associated with t-MDS/AML (98).
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


