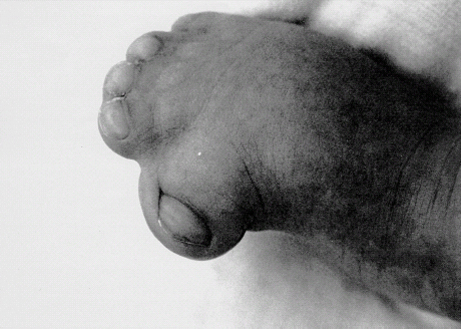
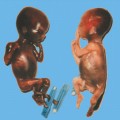
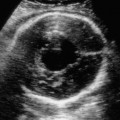
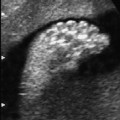
Definition: Also known as acrocephalic syndactyly syndrome. It is characterized by a “tower-shaped” head, facial dysmorphism, and symmetrical syndactyly of the fingers and toes. Incidence: about one in 100 000 births. First described in 1906 by Apert. Etiology/genetics: Partly autosomal-dominant inheritance, but frequently sporadic occurrence (new mutation). Advanced paternal age is a factor favoring its occurrence. Gene defect in the fibroblast growth factor receptor-2 gene (FGFR2), gene locus: 10q26. Clinical features: “Tower-shaped” head, early closure of cranial sutures, anomalies of the cervical vertebral column. Facial anomalies: denting of the forehead in the supraorbital region, hypertelorism, flat orbital bone, exophthalmos, squint, deep-set ears, small, beak-shaped nose, syndactyly (as extreme as “spoon hands”), fusion of the bony parts of fingers II–IV, short fingers, possibly short upper extremities. Mental impairment is not obligatory; in 80% of cases, the intelligence quotient lies above 50–70, and often it is normal. Other anomalies of the cardiac, renal, and gastrointestinal systems may also be present. Ultrasound findings: The earliest prenatal diagnosis was possible at 12 weeks of gestation with nuchal translucency measurements. Syndactyly could also be detected at this stage. At a later stage, an unusual shape of the head resulting from premature closure of the cranial sutures (tower-shaped head) and facial dysmorphism (hypoplasia of the midfacial region) are characteristic features. Detailed scanning may also reveal deep-set ears, unusual shape of the nose, exophthalmos, and hypertelorism. Differential diagnosis: Carpenter syndrome, Crouzon syndrome, cloverleaf skull anomaly, Pfeiffer syndrome, thanatophoric dysplasia, achondrogenesis, Cornelia de Lange syndrome, EEC syndrome, Fryns syndrome, Joubert syndrome, multiple pterygium syndrome, Noonan syndrome, Roberts syndrome, Smith–Lemli–Opitz syndrome, trisomy 21. Clinical management: Further ultrasound screening including fetal echocardiography. Karyotyping (differential diagnosis), molecular-genetic evidence of the defective gene. Fig. 11.1 Apert syndrome. Fetal profile at 22 + 6 weeks. The striking feature is the high and prominent forehead. Self-Help Organization Title: Apert Syndrome Pen Pals Description: Group correspondence program for persons with Apert syndrome to share experience. Information and referrals, pen pals, phone help. Scope: National network Founded: 1992 Address: P.O. Box 115, Providence, RI 02901, United States Telephone: 401–454–0704 (after 4: 30 p.m.) Title: Apert Support and Information Network Description: Provides information and support to families and individuals facing the challenge of Apert syndrome. Provides information and referrals, newsletter, phone support network, pen pals, and annual family get-togethers. Scope: International network Founded: 1995 Address: P.O. Box 1184, Fair Oaks, CA 95628, United States Telephone: 916–961–1092 Fax: 916–961–1092 E-mail: apertnet@ix.netcom.com Fig. 11.2 Apert syndrome. Profile of a child with Apert syndrome. Fig. 11.3 Apert syndrome. Syndactyly of the hand, seen at 22 + 6 weeks. Fig. 11.4 Apert syndrome. Syndactyly of the foot at 22 + 6 weeks. Fig. 11.5 Apert syndrome. Foot of a newborn infant with Apert syndrome. Ferreira JC, Carter SM, Bernstein PS et al. Secondtrimester molecular prenatal diagnosis of sporadic Apert syndrome following suspicious ultrasound findings. Ultrasound Obstet Gynecol 1999; 14: 426–30. Filkins K, Russo IF, Boehmer S et al. Prenatal ultrasonographic and molecular diagnosis of Apert syndrome. Prenat Diagn 1997; 17: 1081–4. Hafner E, Sterniste W, Scholler J, Schuchter K, Philipp K. Prenatal diagnosis of facial malformations. Prenat Diagn 1997; 17: 51–8. Hill LM, Thomas ML, Peterson CS. The ultrasonic detection of Apert syndrome. J Ultrasound Med 1987; 6: 601–4. Kaufmann K, Baldinger S, Pratt L. Ultrasound detection of Apert syndrome: a case report and literature review. Am J Perinatol 1997; 14: 427–30. Lyu KJ, Ko TM. Prenatal diagnosis of Apert syndrome with widely separated cranial sutures. Prenat Diagn 2000; 20: 254–6. Mahieu-Caputo D, Sonigo P, Amiel J et al. Prenatal diagnosis of sporadic Apert syndrome: a sequential diagnostic approach combining three-dimensional computed tomography and molecular biology. Fetal Diagn Ther 2001; 16: 10–2. Narayan H, Scott IV. Prenatal ultrasound diagnosis of Apert’s syndrome. Prenat Diagn 1991; 11: 187–92. Witters I, Devriendt K, Moerman P, van Hole C, Fryns JP. Diaphragmatic hernia as the first echographic sign in Apert syndrome. Prenat Diagn 2000; 20: 404–6. Definition: This is a metabolic dysplasia syndrome with omphalocele, macroglossia, enlargement of organs, giantism, ear malformation, and hypoglycemia in the postnatal stage. There is an increased incidence of blastoma in childhood, especially Wilms tumor and adrenocortical cancers. Incidence: One in 12000–50 000 births. First described in 1965 by Beckwith and Wiedemann. Clinical history/genetics: Heterogeneous syndrome. Gene locus: 11 p15.5. The cause is dysregulation of a parental gene (genomic imprinting), which affects growth. Eightyfive percent of cases are sporadic, and 15% are of autosomal-dominant inheritance. In some cases of dominant inheritance, mutation of the cyclin-dependent kinase inhibitor 1C (p57, Kip2) gene (CDKN1 C) is present. In the sporadic form, chromosomal aberrations are present in 1% of cases. Another 10–20% show paternal disomy 11; this means that the 11 p15 region of both chromosomes originates from the father and that maternal alleles are not present. Some patients demonstrate mutation of the KVLQT1 gene and, as in the case of dominant inheritance, of the CDKN1 C gene. In 50% of cases, an imprinting mutation is seen in which expression of the insulin-like growth factor-2 gene (IGF2) is also determined by a maternal allele. Ultrasound findings: Macrosomia is evident in the later stages of the second trimester. Other features are: hydramnios, anomalies of the placenta such as partial molar degeneration, macroglossia, hypertrophy of organs, especially large and echogenic kidneys, cardiomyopathy and cardiomegaly, and omphalocele. In addition, cryptorchism and hypospadias may also be evident. Detection has been reported as early as at 12 weeks of gestation. Differential diagnosis: Diabetic fetopathy, Perlman syndrome, Simpson–Golabi–Behmel syndrome, Marfan syndrome, Marshall–Smith syndrome, Sotos syndrome, Weaver syndrome. Prognosis: The perinatal fatality is about 20% and is due to macrosomia, omphalocele, hypoglycemia, fits, and cardiac failure. Mental development may be adequate as long as damage to the brain tissue due to hypoglycemia can be avoided (the tendency to hypoglycemia can continue into the first years of life). Hypertrophy of the viscera regresses in the course of development. Body size may be normal. Regular checks are necessary in the first 6 years of life for early detection of blastomas. Self-Help Organization Title: Beckwith–Wiedemann Support Network Description: Support and information for parents of children with Beckwith–Wiedemann syndrome or isolated hemihypertrophy, and interested medical professionals. Newsletter, parent directory, information and referrals, phone support. Aims to increase public awareness and encourages research. Scope: International network Founded: 1989 Address: 2711 Colony Rd., Ann Arbor, MI 48104, United States Telephone: 734–973–0263; parents’ toll-free number 1–800–837–2976 Fax: 734–973–9721 E-mail: a800bwsn@aol.com Cobellis G, Iannoto P, Stabile M et al. Prenatal ultrasound diagnosis of macroglossia in the Wiedemann–Beckwith syndrome. Prenat Diagn 1988; 8: 79–81. Fert-Ferrer S, Guichet A, Tantau J et al. Subtle familial unbalanced translocation t (8; 11)(p23.2;p15.5) in two fetuses with Beckwith–Wiedemann features. Prenat Diagn 2000; 20: 511–5. Fremond B, Poulain P, Odent S, Milon J, Treguier C, Babut JM. Prenatal detection of a congenital pancreatic cyst and Beckwith–Wiedemann syndrome. Prenat Diagn 1997; 17: 276–80. Hamada H, Fujiki Y, Obata-Yasuoka M, Watanabe H, Yamada N, Kubo T. Prenatal sonographic diagnosis of Beckwith–Wiedemann syndrome in association with a single umbilical artery. J Clin Ultrasound 2001; 29: 535–8. Hewitt B, Bankier A. Prenatal ultrasound diagnosis of Beckwith–Wiedemann syndrome. Aust N Z J Obstet Gynaecol 1994; 34: 488–90. Nowotny T, Bollmann R, Pfeifer L, Windt E. Beckwith–Wiedemann syndrome: difficulties with prenatal diagnosis. Fetal Diagn Ther 1994; 9: 256–60. Reish O, Lerer I, Amiel A et al. Wiedemann–Beckwith syndrome: further prenatal characterization of the condition [review]. Am JMed Genet 2002; 107: 209–13. Shah YG, Metlay L. Prenatal ultrasound diagnosis of Beckwith–Wiedemann syndrome. J Clin Ultrasound 1990; 18: 597–600. Viljoen DL, Jaquire Z, Woods DL. Prenatal diagnosis in autosomal-dominant Beckwith–Wiedemann syndrome. Prenat Diagn 1991; 11: 167–75. Whisson CC, Whyte A, Ziesing P Beckwith–Wiedemann syndrome: antenatal diagnosis. Australas Radiol 1994; 38: 130–1. Winter SC, Curry CJ, Smith JC, Kassel S, Miller L, Andrea J. Prenatal diagnosis of the Beckwith–Wiedemann syndrome. Am J Med Genet 1986; 24: 137–41. Definition: A fatal malformation, probably resulting from early rupture of the amnion. At least two of three malformations must be present as diagnostic criteria: myelomeningocele or caudal regression syndrome, thoraco-abdominoschisis or abdominoschisis, anomalies of the extremities. Incidence: One in 14 000 births. Clinical history: Sporadic occurrence. Teratogen: Not known. Origin: Early developmental disturbances in the embryonic stage due to early amnion rupture have been discussed as a possible cause. Part of the embryo may be found in the celom. Ultrasound findings: Following are characteristic findings: large abdominal wall defects, severe neural tube defects, extreme kyphoscoliosis with shortening of the vertebral column and caudal regression syndrome. A typical feature is an absence of one or more extremities. Club foot may also be present. Meningomyelocele may be associated with Arnold–Chiari malformation and development of hydrocephalus. Ectopia cordis may result from thoraco-abdominoschisis. In addition, facial clefts are observed. In extreme cases, the umbilical cordmay even be absent, the fetus being joined directly to the placenta. Increased nuchal translucency is also a typical feature. Clinical management: Karyotyping to evaluate the risk of recurrence (translocation?). Determination of the extent of malformations for further prognosis. Termination of pregnancy is justified in most cases. Obstetric intervention due to fetal distress should be avoided. Fig. 11.6 Body stalk anomaly. Longitudinal section of the lower torso at 9 + 3 weeks, with an extensive omphalocele-like structure. Fig. 11.7 Body stalk anomaly. Demonstration of the fetal head within the amnion as well as an omphalocele-like structure outside the amnion in the extraembryonic celom, in a case of body stalk anomaly at 9 + 3 weeks. Procedure after birth: Intensive medical treatment of the neonate should be avoided. Prognosis: In case of full expression of the anomaly, the outcome is fatal. Only mildly affected individuals may survive. See also the section (8.4 above) on amniotic band syndrome. Becker R, Runkel S, Entezami M. Prenatal diagnosis of body stalk anomaly at 9 weeks of gestation. Fetal diagnosis and therapy 2000; 15: 301–3. Chang Y, Silverman N, Jackson L, Wapner R, Wallerstein R. Maternal uniparental disomy of chromosome 16 and body stalk anomaly. Am J Med Genet 2000; 94: 284–6. Ginsberg NE, Cadkin A, Strom C. Prenatal diagnosis of body stalk anomaly in the first trimester of pregnancy. Ultrasound Obstet Gynecol 1997; 10: 419–21. Hiett AK, Devoe LD, Falls DG, Martin SA. Ultrasound diagnosis of a twin gestation with concordant body stalk anomaly: a case report. J Reprod Med 1992; 37: 944–6. Jauniaux E, Vyas S, Finlayson C, Moscoso G, Driver M, Campbell S. Early sonographic diagnosis of body stalk anomaly. Prenat Diagn 1990; 10: 127–32. Martinez JM, Fortuny A, Comas C et al. Body stalk anomaly associated with maternal cocaine abuse. Prenat Diagn 1994; 14: 669–72. Paul C, Zosmer N, Jurkovic D, Nicolaides K. A case of body stalk anomaly at 10 weeks of gestation. Ultrasound Obstet Gynecol 2001; 17: 157–9. Shalev E, Eliyahu S, Battino S, Weiner E. First trimester transvaginal sonographic diagnosis of body stalk anomaly [correction of anatomy]. J Ultrasound Med 1995; 14: 641–2. Definition: CHARGE is the acronym for coloboma, heart disease, atresia of choanae, retarded mental development, genital hypoplasia, ear anomalies and deafness. Incidence: Rare; only 200 cases have been described. Origins/genetics: Mostly sporadic occurrence (multifactorial inheritance), occasionally autosomal-dominant. The cause appears to be a disturbance in differentiation at the embryonic stage between the 35th and 45th day after conception. In sporadic forms, the risk of recurrence is 1%. Clinical features: Mental impairment, ocular coloboma; rarely, microphthalmia and anophthalmia are present. The choanal atresia is usually bilateral. Dysplasia of the auricles, hearing impairment, and deafness may also be detected. Typical cardiac anomalies consist of persistent Botallo’s duct, ASD, VSD, tetralogy of Fallot, and AV canal. Seventy-four percent of cases show hypoplasia of the genitals. In addition, growth retardation, facial clefts, atresia of the esophagus, renal malformations, microgenia, hemivertebrae, syndactyly, scoliosis, facial pareses, central nervous system anomalies, and anal atresia may also be detected. Fig. 11.8 CHARGE association. Cross-section of fetal head at 18 + 1 weeks: widening of both the lateral ventricles, underdeveloped and coil-shaped choroid plexus. Ultrasound findings: Accurate classification of this syndrome at the prenatal stage is often difficult. The diagnosis is confirmed if the following combination of anomalies is detected: severe hydramnios, enlargement of cerebral ventricles, Dandy–Walker variant, genital hypoplasia, small stomach and cardiac anomaly, possibly dysplasia of the ear. Suspicion of choanal atresia can be verified using color-coded Doppler imaging. Differential diagnosis: Wolf–Hirschhorn syndrome, trisomy 13, trisomy 18, Noonan syndrome, Pena–Shokeir syndrome, Smith–Lemli–Opitz syndrome, Treacher–Collins syndrome, VACTERL association. Prognosis: This depends on the extent of the malformations. In severe cases, death results soon after birth, due either to respiratory failure, treatment-resistant hypocalcemia, or heart failure. Mental impairment can be expected in survivors. Self-Help Organization Title: CHARGE Syndrome Foundation, Inc. Description: Networking of families affected by CHARGE syndrome. Offers an organization manual for parents, newsletter, information, and parent-to-parent support. Biannual international conference. Newsletter. Scope: National network Founded: 1993 Address: 2004 Parkade, Columbia, MO 65202, United States Telephone: 573–499–4694. Families only call 1–800–442–7604 Fax: 573–499–4694 E-mail: marion@chargesyndrome.org Becker R, Stiemer B, Neumann L, Entezami M. Mild ventriculomegaly, mild cerebellar hypoplasia and dysplastic choroid plexus as early prenatal signs of CHARGE association. Fetal Diagn Ther 2001; 16: 280–3. Hertzberg BS, Kliewer MA, Lile RL. Antenatal ultrasonographic findings in the CHARGE association. J Ultrasound Med 1994; 13: 238–42. Definition: This syndrome consists of malformations of the face and the extremities, growth retardation, microcephaly, and mental impairment. Incidence: On the whole, relatively rare; 300 cases have been described. First described by Brachmann in 1916 and de Lange in 1933. Origin/genetics: Isolated autosomal-dominant or autosomal-recessive inheritance, with variable expression. Gene locus: 3q26.3; mostly sporadic occurrence. Ultrasound findings: An abnormal nuchal translucency is detected in the first trimester. Growth restriction is first evident at 20–25 weeks. In addition, brachycephaly, microcephaly, and microgenia are diagnosed. Cardiac anomalies such as VSD and ASD are common. Further anomalies include hypospadias, cryptorchism, micromelia, syndactyly, dysplasia of ulna, oligodactyly. The earliest diagnosis was made at 12 weeks on the basis of a nuchal edema. Differential diagnosis: Apert syndrome, chromosomal aberrations, Fanconi anemia, Holt–Oram syndrome, multiple pterygium syndrome, Roberts syndrome, Smith–Lemli–Opitz syndrome, TAR syndrome. Clinical management: Further ultrasound screening, including fetal echocardiography, karyotyping (normal finding). A low level of alpha fetoprotein in maternal serum has been reported in association with Cornelia de Lange syndrome. Prognosis: The perinatal mortality is increased. Survivors show mental impairment and growth restriction. Self-Help Organization Title: Cornelia de Lange Syndrome Foundation, Inc. Description: Provides support and education to families affected by Cornelia de Lange syndrome. Supports research. Newsletter. Family album available for networking and mutual support. Annual convention. Professional network. Assistance in starting groups. Scope: International Number of groups: 2500 + member families Founded: 1981 Address: 302 West Main St., Suite 100, Avon, CT 6001, United States Telephone: 1–800–223–8355 or 860–676–8166 Fax: 860–676–8337 E-mail: CDLSintl@iconn.net Boog G, Sagot F, Winer N, David A, Nomballais MF. Brachmann–de Lange syndrome: a cause of early symmetric fetal growth delay. Eur J Obstet Gynecol Reprod Biol 1999; 85: 173–7. Bruner JP, Hsia YE. Prenatal findings in Brachmann–de Lange syndrome. Obstet Gynecol 1990; 76: 966–8. Huang WH, Porto M. Abnormal first-trimester fetal nuchal translucency and Cornelia de Lange syndrome. Obstet Gynecol 2002; 99: 956–8. Kliewer MA, Kahler SG, Hertzberg BS, Bowie JD. Fetal biometry in the Brachmann–de Lange syndrome. Am J Med Genet 1993; 47: 1035–41. Marino T, Wheeler PG, Simpson LL, Craigo SD, Bianchi DW. Fetal diaphragmatic hernia and upper limb anomalies suggest Brachmann–de Lange syndrome. Prenat Diagn 2002; 22: 144–7. Sekimoto H, Osada H, Kimura H, Kamiyama M, Arai K, Sekiya S. Prenatal findings in Brachmann–de Lange syndrome. Arch Gynecol Obstet 2000; 263: 182–4. Urban M, Hartung J. Ultrasonographic and clinical appearance of a 22-week-old fetus with Brachmann–de Lange syndrome. Am J Med Genet 2001; 102: 73–5. Definition: Premature closure of the cranial sutures, causing characteristic anomalies of the head and face. The coronal, lambdoid, and sagittal sutures are mainly affected. The resulting dysmorphisms include a clover-shaped or tower-shaped skull, hypoplasia of the midfacial region, and exophthalmos. First described in 1912 by Crouzon. Origin/genetics: Autosomal-dominant inheritance. Mutation of the fibroblast growth factor receptor-2 gene. Gene locus: 10 q25–q26. New mutations are observed in about 25% of cases. Ultrasound findings: These include an abnormally shaped skull, in severe cases clover-shaped skull (which is a special form of tower-shaped head), hypertelorism, beaked nose, micro-gnathia, progenia, exophthalmos, possibly combined with widening of the cerebral ventricles, agenesis of the corpus callosum, and cleft lip and palate. If there is a positive family history, it is possible to diagnose this disorder in the second trimester on finding widening of the interorbital distance. Differential diagnosis: Apert syndrome, Carpenter syndrome, clover-shaped skull, Pfeiffer syndrome, thanatophoric dysplasia. Procedure after birth: Surgery is necessary due to closure of the sutures. Some of the skull deformities can be corrected in this way. Mental development is improved after neurosurgical interventions to reduce intracranial pressure. Prognosis: Atrophy of the optical nerve may cause blindness. Respiratory obstruction is also possible. Fits and mental impairment are rarely seen. Self-Help Organization Title: Craniosynostosis and Parents Support, Inc. Description: Dedicated to helping families find support and information to help deal with craniosynostosis. The aim is to raise awareness through public and professional education. Provides a newsletter, literature, hospital care packages, pen pals, and phone support. Advocacy, information and referrals. Supports research. Scope: International network Founded: 1999 Address: 1136 Iris Lane, Beaufort, SC 29906, United States Telephone: 1–877–686-CAPS Fax: 843–846–0779 E-mail: CAPS2000@gosiggy.com Kjaer I, Hansen BF, Kjaer KW, Skovby F. Abnormal timing in the prenatal ossification of vertebral column and hand in Crouzon syndrome. Am J Med Genet 2000; 90: 386–9. Leo MV, Suslak L, Ganesh VL, Adhate A, Apuzzio JJ. Crouzon syndrome: prenatal ultrasound diagnosis by binocular diameters. Obstet Gynecol 1991; 78: 906–8. Martinelli P, Russo R, Agangi A, Paladini D. Prenatal ultrasound diagnosis of frontonasal dysplasia. Prenat Diagn 2002; 22: 375–9. Miller C, Losken HW, Towbin R et al. Ultrasound diagnosis of craniosynostosis. Cleft Palate Craniofac J 2002; 39: 73–80. Definition: Short-limb dwarfism with hexadactyly, hypoplasia of the nails, abnormal connection of the mucous membranes between the upper lip and upper gum. Incidence: Rare; about 250 cases have been reported; there is an incidence of one in 200 in the Amish population (Lancaster County, Pennsylvania, USA). First described in 1940. Ultrasound findings: Disproportionate short-limb dwarfism, with shortening of the limbs increasing distally. Hexadactyly of the hands, occasionally of the feet. Short femur, club feet, Dandy–Walker malformation, sometimes genital anomalies. Cardiac anomalies are found in 50% of cases, most frequently ASD. Differential diagnosis: At the neonatal stage, short rib-polydactyly syndrome has to be differentiated from this. Definitive diagnosis is possible at a later stage. Clinical management: If there is a positive family history, chorionic villus sampling and DNA analysis is recommended. Prognosis: There is an increased neonatal mortality due to respiratory complications resulting from short ribs (30–50% of affected infants). In addition, the prognosis also depends on the associated cardiac anomaly. The adult height varies considerably, between 105 and 160 cm. Affected individuals have normal intelligence. Arya L, Mendiratta V, Sharma RC, Solanki RS. Ellis–van Creveld Syndrome: a report of two cases. Pediatr Dermatol 2001; 18: 485–9. Berardi JC, Moulis M, Laloux V, Godard J, Wipff P, Botto C. Ellis–van Creveld syndrome: contribution of echo-graphy to prenatal diagnosis–a propos of a case. J Gynecol Obstet Biol Reprod (Paris) 1985; 14: 43–7. Dugoff L, Thieme G, Hobbins JC. First trimester prenatal diagnosis of chondroectodermal dysplasia (Ellis–van Creveld syndrome) with ultrasound. Ultrasound Obstet Gynecol 2001; 17: 86–8. Guschmann M, Horn D, Gasiorek-Wiens A, Urban M, Kunze J, Vogel M. Ellis–van Creveld syndrome: examination at 15 weeks’ gestation. Prenat Diagn 1999; 19: 879–83. Meinel K, Himmel D. Status of ultrasound and roentgen diagnosis in prenatal detection of osteochondrodys-plasias. Zentralbl Gynäkol 1987; 109: 1303–3. Sergi C, Voigtlander T, Zoubaa S et al. Ellis–van Creveld syndrome: a generalized dysplasia of enchondral ossification. Pediatr Radiol 2001; 31: 289–93. Tongsong T, Chanprapaph P. Prenatal sonographic diagnosis of Ellis–van Creveld syndrome. J Clin Ultrasound 2000; 28: 38–41. Torrente I, Mangino M, De Luca A et al. First-trimester prenatal diagnosis of Ellis–van Creveld syndrome using linked microsatellite markers. Prenat Diagn 1998; 18: 504–6. Zimmer EZ, Weinraub Z, Raijman A, Pery M, Peretz BA. Antenatal diagnosis of a fetus with an extremely narrow thorax and short limb dwarfism. JCU J Clin Ultrasound 1984; 12: 112–4. Definition: This is a syndrome with a characteristic facial expression of “whistling face,” hypoplasia of the nostrils, ulnar deviation of the hands, bent fingers and club feet. Incidence: Generally rare; only 100 cases have been reported. Origin/genetics: Autosomal-dominant inheritance is the frequent form, although autosomal-recessive and sporadic forms have also been described. Clinical features: A rounded, chubby, mask-like face without expression; the small mouth appears to be “whistling.” Ultrasound findings: Hypertelorism, deep-set nasal bridge, small nose, hypoplasia of the nostrils, long median ridge in the upper lip, ulnar deviation of the hands, contracted bent fingers, especially the thumb, and club feet with contractures of the toes. Raised palate, possibly cleft palate. Short neck with pterygium. Dwarfism, scoliosis, microcephaly. In a case with a positive family history, the diagnosis was made at 21 weeks due to club feet and facial anomalies. Differential diagnosis: Arthrogryposis, Schwartz–Jampel syndrome, amniotic band sequence, Cornelia de Lange syndrome, multiple pterygium syndrome, Pena–Shokeir syndrome, Seckel syndrome, Smith–Lemli–Opitz syndrome, trisomy 18. Prognosis: Life expectancy is normal, usually with no mental impairment. However, cases of mental impairment and fits have also been described. Title: Freeman–Sheldon Parent Support Group Description: Emotional support for parents of children with Freeman–Sheldon and for adults with this syndrome. Sharing of helpful medical literature library. Information on growth and development of individuals affected. Participates in research projects. Members network by phone and mail. Newsletter. Scope: International network Founded: 1982 Address: 509E. Northmont Way, Salt Lake City, UT 84103, United States Telephone: 801–364–7060 E-mail: fspsg@aol.com Web: http://www.fspsg.org Cruickshanks GF, Brown S, Chitayat D. Anesthesia for Freeman–Sheldon syndrome using a laryngeal mask airway. Can J Anaesth 1999; 46: 783–7. Krakowiak PA, O’Quinn JR, Bohnsack JF et al A variant of Freeman–Sheldon syndrome maps to 11 p15.5-pter. Am J Hum Genet 1997; 60: 426–32. Krakowiak PA, Bohnsack JF, Carey JC, Bamshad M. Clinical analysis of a variant of Freeman–Sheldon syndrome (DA2B). Am J Med Genet 1998; 76: 93–8. Manji KP, Mbise RL. Generalized muscle hypertonia with mask-like face (Freeman–Sheldon syndrome) in a Tanzanian girl. Clin Genet 1998; 54: 252–3. Robbins-Furman P, Hecht JT, Rocklin M, Maklad N, Greenhaw G, Wilkins I. Prenatal diagnosis of Freeman–Sheldon syndrome (whistling face). Prenat Diagn 1995; 15: 179–82. Robinson PJ. Freeman–Sheldon syndrome: severe upper airway obstruction requiring neonatal tracheostomy [review]. Pediatr Pulmonol 1997; 23: 457–9. Schefels J, Wenzl TG, Merz U et al. Functional upper airway obstruction in a child with Freeman–Sheldon syndrome. ORL J Otorhinolaryngol Relat Spec 2002; 64: 53–6. Schrander-Stumpel C, Fryns JP, Beemer FA, Rive FA. Association of distal arthrogryposis, mental retardation, whistling face, and Pierre Robin sequence: evidence for nosologic heterogeneity. Am J Med Genet 1991; 38: 557–61. Definition: This syndrome combines facial dysmorphism, anomalies of the distal extremities, and diaphragmatic hernia. First described in 1979 by Fryns. Incidence: About one in 15 000 births. Origin/genetics: Autosomal-recessive inheritance. Ultrasound findings: Thickening of the neck region in the form of hygroma colli is evident in the first trimester. Later on, hydramnios and fetal hydrops develop. Craniofacial dysmorphism is characterized by coarse facial features, a prominent glabella, flat nasal bridge, large nose with anteverted nostrils, short upper lip, macrostomia, cleft lip and palate, retrogenia, and dysplasia of the auricles. A characteristic feature is diaphragmatic hernia, the posterolateral part being absent. Malformations of the limbs are also present: brachytelephalangia (hypoplasia of the distal phalanges) of the fingers and toes. In addition, microphthalmia, clouded cornea, retinal dysplasia, short neck, pterygium, cystic renal dysplasia, Dandy–Walker malformation, widening of the cranial ventricles, club feet, cardiac defects such as VSD, and anal atresia may also be detected. Diagnosis is possible at the earliest at 13 weeks due to severe hygroma colli. Differential diagnosis: Hydrolethalus, Schinzel–Giedion syndrome, Rüdiger syndrome, Wolf–Hirschhorn syndrome, tetrasomy 12p, trisomy 18 and 21, Walker–Warburg syndrome, achondrogenesis, Apert syndrome, Cornelia de Lange syndrome, Kniest syndrome, multiple pterygium syndrome, Noonan syndrome, Roberts syndrome, Smith–Lemli–Opitz syndrome. Prognosis: There is a high mortality rate due to respiratory failure (pulmonary hypoplasia) secondary to diaphragmatic hernia. In the absence of the above, infants may survive, but mental impairment can be expected. Fig. 11.9 Fryns syndrome. Fetus at 12 + 3 weeks with severe cystic hygroma, nuchal thickness measuring 8 mm. Chorionic villus sampling showed a normal karyotype. Fig. 11.10 Fryns syndrome. Same fetus. Cross-section of the fetal neck. Further clinical progress: normal fetal echocardiography, regression of hygroma. Fig. 11.11 Fryns syndrome. Same fetus seen at 36 + 3 weeks: for the first time, the heart is seen displaced to the right (lv: left ventricle), due to the left kidney (arrows) being pushed up into the thoracic cavity: fetal diaphragmatic hernia. Fryns syndrome was diagnosed after birth. Dix U, Beudt U, Langenbeck U. Fryns syndrome: pre- and postnatal diagnosis. Z Geburtshilfe Perinatol 1991; 195: 280–4. Enns GM, Cox VA, Goldstein RB, Gibbs DL, Harrison MR, Golabi M. Congenital diaphragmatic defects and associated syndromes, malformations, and chromosome anomalies: a retrospective study of 60 patients and literature review. Am J Med Genet 1998; 79: 215–25. Hösli IM, Tercanli S, Rehder H, Holzgreve W. Cystic hygroma as an early first-trimester ultrasound marker for recurrent Fryns’ syndrome. Ultrasound Obstet Gynecol 1997; 10: 422–4. Meinecke P, Fryns JP. The Fryns syndrome: diaphragma; tic defects, craniofacial dysmorphism, and distal digital hypoplasia: further evidence for autosomal-recessive inheritance. Clin Genet 1985; 28: 516–20. Ramsing M, Gillessen-Kaesbach G, Holzgreve W, Fritz B, Rehder H. Variability in the phenotypic expression of Fryns syndrome: a report of two sibships. Am J Med Genet 2000; 95: 415–24. Sheffield JS, Twickler DM, Timmons C, Land K, Harrod MJ, Ramus RM. Fryns syndrome: prenatal diagnosis and pathologic correlation. J Ultrasound Med 1998; 17: 585–9. Van Wymersch D, Favre R, Gasser B. Use of three-dimensional ultrasound to establish the prenatal diagnosis of Fryns syndrome. Fetal Diagn Ther 1996; 11: 335–40. Definition: This is a group of malformations consisting of facial asymmetry, anomalies of the eyes, ears and cheeks. It may also be combined with defects in the vertebral column. Incidence: One in 3000–5000 births. Origin/genetics: This is a heterogeneous and complex malformation syndrome. The occurrence is mostly sporadic—for example, due to restricted placenta perfusion at a sensitive stage of development. There is a disturbance in the development of the first and second branchial arches. Rarely, autosomal-dominant or autosomal-recessive inheritance has been observed. Males are more often affected. With one affected child, there is a 3% risk of recurrence; if two siblings are affected, the risk rises to 6%. Clinical features and ultrasound findings: Asymmetrical face resulting from unilateral hypoplasia of bone and soft tissue, frontal bossing, hypoplasia of zygomatic arch and mandibular bone, retrogenia. Malformations of the eye: epibulbar dermoid or lipodermoid at the junction of the sclera and cornea, coloboma of the upper lid. Malformations of the ear: preauricular tags, small ears, anomalies of the external ear. Further anomalies include: unilateral microstomia, cleft jaw, partial vertebral scoliosis. Development of hydramnios is frequent. In addition, cardiac defects, renal anomalies and widening of the cranial ventricles are also detected. The main finding is slight facial dysmorphism and hydramnios. The earliest diagnosis was reported at 16 weeks on the basis of maxillary clefts and unilateral microphthalmia. Associated syndromes: Mandibulofacial dysostosis, hemifacial microsomia, Wildervanck syndrome, Frase syndrome, Fryns syndrome, Klippel–Pfeil sequence, Nager syndrome, Treacher–Collins syndrome, trisomy 13 and 18. Prognosis: This is usually favorable, with normal mental development and intelligence. Cosmetic surgery is needed to correct the facial anomalies. Physical handicap may result due to deafness and microphthalmia. Self-Help Organization Title: Goldenhar Syndrome Support Network Description: Support and information for families affected by Goldenhar syndrome (hemifacial microsomia). Information and referrals, newsletter, literature, pen pals, and advocacy. Online mailing list. Scope: International network Founded: 1998 Address: 9325 163 Street, Edmonton, Alberta T5R 2P4, Canada E-mail: bbds.page@i.am De Catte L, Laubach M, Legein J, Goossens A. Early prenatal diagnosis of oculoauriculovertebral dysplasia or the Goldenhar syndrome. Ultrasound Obstet Gynecol 1996; 8: 422–4. Tamas DE, Mahony BS, Bowie JD, Woodruff WW HH. Prenatal sonographic diagnosis of hemifacial microsomia (Goldenhar–Gorlin syndrome). J Ultrasound Med 1986; 5: 461–3. Witters I, Schreurs J, Van Wing J, Wouters W, Fryns JP. Prenatal diagnosis of facial clefting as part of the oculo-auriculo-vertebral spectrum. Prenat Diagn 2001; 21: 62–4. Definition: This syndrome consists of anomalies of the heart and upper limbs. Incidence: Rare, although over a hundred cases have been reported. First described in 1960 by Holt and Oram. Origin/genetics: Autosomal-dominant inheritance with full penetrance and variable expression. New mutations are found in 50–80%. Gene locus: 12q24.1. Girls are more severely affected than boys. Ultrasound findings: Cardiac anomalies (85%): ASDII (partially combined with cardiac arrhythmia); rarely VSD. Malformations of the upper extremities: three digits or hypoplasia or aplasia of the thumb, dysplasia or aplasia of the radius, and anomalies of the small finger, elbow, and shoulder joint. Occasionally, radioulnar synostosis, hypoplasia of the humerus, and phocomelia have also been observed. Diagnosis is possible in the second trimester after careful scanning of the lower arm and thumb. Differential diagnosis: TAR syndrome, Fanconi anemia, thalidomide syndrome, Tabatznik syndrome, Rogers syndrome, Cornelia de Lange syndrome, Nager syndrome, trisomy 13 and 18, VACTERL association. Prognosis: This depends on the type of cardiac anomaly present; normal mental development can be expected. Brons JT, van Geijn HP, Wladimiroff JW et al. Prenatal ultrasound diagnosis of the Holt–Oram syndrome. Prenat Diagn 1988; 8: 175–81. Lehner R, Wenzl R, Vanura H, Frank W, Safar P, Husslein P. Diagnosis of familial Holt–Oram syndrome. Z Geburtshilfe Perinatol 1994; 198: 143–9. Muller LM, De Jong G, Van Heerden KM. The antenatal ultrasonographic detection of the Holt–Oram syndrome. S Afr Med J 1985; 68: 313–5. Tongsong T, Chanprapaph P. Prenatal sonographic diagnosis of Holt–Oram syndrome. J Clin Ultrasound 2000; 28: 98–100. Definition: This is a fatal complex of malformations, with hydrocephalus and other cranial anomalies, facial dysmorphism, including facial clefts, polydactyly of the hands and feet, and pulmonary hypoplasia. Incidence: In Finland, one in 20000 births. Incidences in African and Arabic families has also been reported. About 60 cases have been described. First description: 28 cases were reported in Finland in 1981. Genetics: Autosomal-recessive inheritance. Ultrasound findings: Growth restriction, hydramnios (obligatory finding). Head and face: hydrocephalus (93%), agenesis of the corpus callosum, absence of the septum pellucidum, Dandy–Walker malformation; cleft palate, cleft lip (56%), microgenia (84%), small tongue, which may even be absent. Thorax: tracheobronchial malformations (65%), abnormal pulmonary lobes; cardiac defects (58%): VSD, AV canal. Extremities: polydactyly (88%), club feet. Diagnosis is possible as early as at 13 weeks (central nervous system anomalies with limb defects). Hydramnios and severe obstructive hydrocephalus develop at a later stage. In contrast to Meckel–Gruber syndrome, cystic kidneys and encephalocele are absent. Differential diagnosis: Meckel–Gruber syndrome, septooptic dysplasia, Walker–Warburg syndrome, Goldenhar syndrome, Fryns syndrome, Ivemark syndrome, Smith–Lemli–Opitz syndrome, trisomy 13, Apert syndrome, Fanconi anemia, Mohr syndrome, Pfeiffer syndrome, camptomelic dysplasia, Crouzon syndrome, Gorlin syndrome, multiple pterygium syndrome, Neu–Laxova syndrome, Roberts syndrome. Prognosis: The outcome is always fatal, usually in the neonatal stage. Definition: The caudal aplasia–dysplasia sequence is caused by a partial or total absence of the distal part of the neural tube. This results in anomalies of the lower limb and of the gastrointestinal and urogenital tracts. Incidence: One in 20 000–100 000 births. Clinical history/genetics: Mostly sporadic occurrence, genetically heterogeneous. Autosomal or X-chromosome-dominant inheritance has rarely been described. Teratogen: Diabetes mellitus. Embryology: The lower segment of the spinal canal develops until 7 weeks after conception. Primary absence of this segment, rather than secondary regression of an already formed segment, is the most probable cause of the caudal regression syndrome. It is not yet clear whether sirenomelia represents the most severe form of caudal regression syndrome or whether it is a distinct entity. An early insult to the developing lower spinal column results in aplasia or dysplasia of the sacrum. Ultrasound findings:
11 Selected Syndromes and Associations
Apert Syndrome
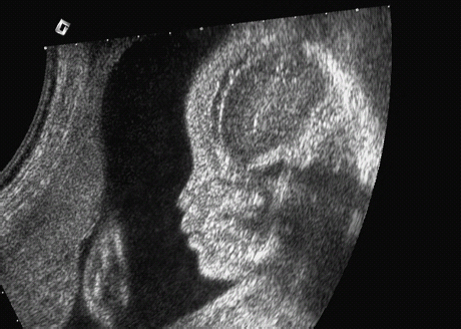
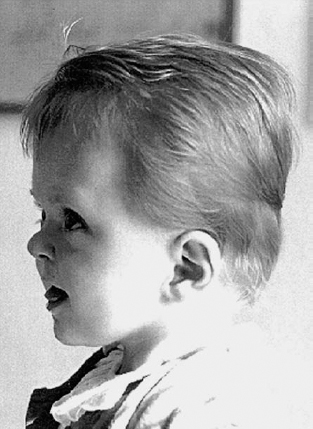
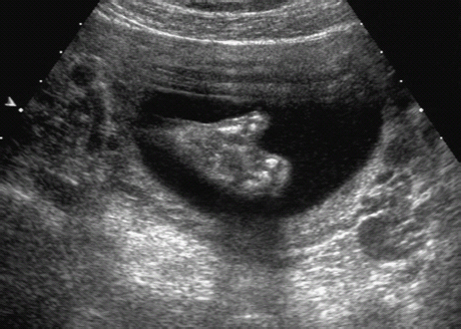
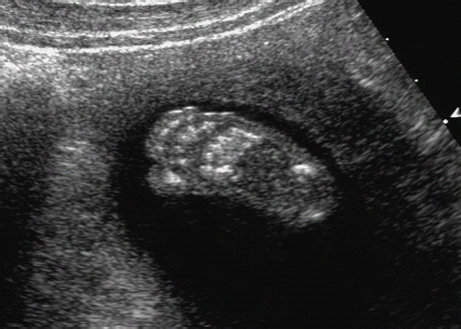
References
Beckwith–Wiedemann Syndrome
References
Body Stalk Anomaly
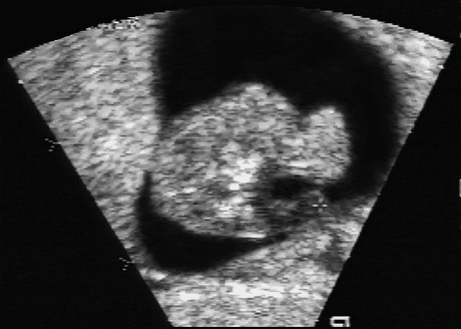
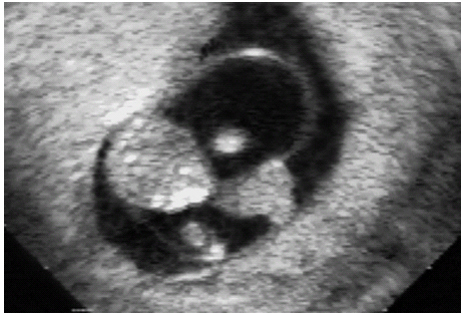
References
CHARGE Association
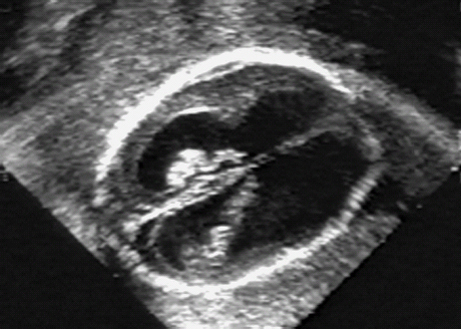
References
Cornelia de Lange Syndrome (Brachmann–de Lange Syndrome)
References
Crouzon Syndrome (Craniofacial Dysostosis Type I)
References
Ellis–van Creveld Syndrome
References
Freeman–Sheldon Syndrome (Whistling Face)
References
Fryns Syndrome
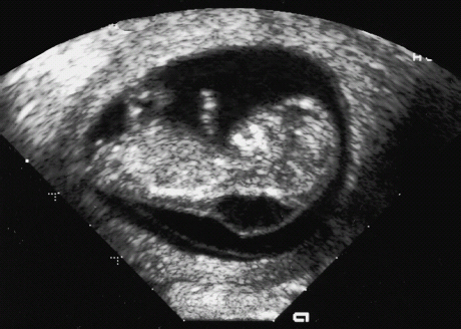
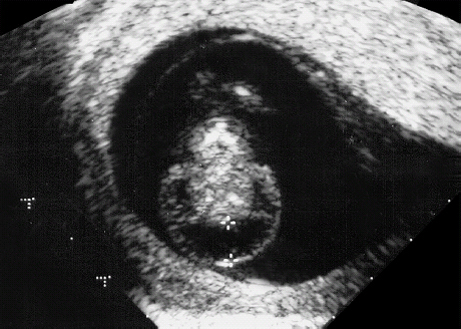
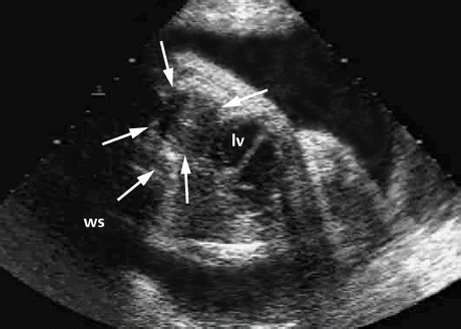
References
Goldenhar Syndrome
References
Holt–Oram Syndrome
References
Hydrolethalus
Caudal Regression Syndrome
![]()
Stay updated, free articles. Join our Telegram channel


Full access? Get Clinical Tree


