Abstract
The short-rib polydactyly syndromes (SRPS) are a group of autosomal recessively inherited skeletal disorders termed skeletal ciliopathies . Ultrasound abnormalities include small, long, narrow chests; shortened appendicular long bones; and frequently seen polydactyly. Visceral abnormalities are commonly seen and occur in the neurologic, cardiac, gastrointestinal, and genitourinary systems, further complicating outcomes. The short-rib polydactyly syndromes, including asphyxiating thoracic dystrophy, are highly associated with lethality because of pulmonary hypoplasia and respiratory compromise.
Keywords
short-rib polydactyly syndromes, Jeune syndrome, cilia
Introduction
The short-rib polydactyly syndromes (SRPSs) are a genetically heterogeneous group of autosomal recessive skeletal disorders. Characteristic findings in SRPS include short horizontal ribs, shortened appendicular long bones limbs, and polydactyly. Historically, four distinct SRPS types have been recognized :
- •
SRPS I (Saldino-Noonan syndrome)
- •
SRPS II (Majweski syndrome)
- •
SRPS III (Verma-Naumoff syndrome)
- •
SRPS IV (Beemer-Langer syndrome)
Disorder
Definition
SRPSs are defined by radiographic findings that include long narrow thoraces with horizontal ribs, shortened appendicular bones, often associated with hypoplastic or poorly formed bones, polydactyly, and frequently other concomitant organ system abnormalities. Similar to other Mendelian disorders that result from alterations in cilia function, multiorgan system abnormalities are common.
Prevalence and Epidemiology
Because of the rarity of this disorder, the exact incidence is unknown, though it is estimated that the frequency is 1 : 100,000 to 1 : 130,000 live births. It is well established that these disorders are autosomal recessive.
Etiology
SRPS transmission is autosomal recessive. All four SRPS types and Jeune syndrome have considerable overlap. Molecular analysis has shown both allelic and locus heterogeneity; thus mutations in the same gene can produce a wide spectrum of phenotype from immediate newborn lethality to long-term survivors.
The SRPS disorders share a narrow thorax, micromelia, polydactyly, and frequently visceral abnormalities. Jeune syndrome or ATD is very similar to SRPS type III; however, the clinical and radiographic findings are somewhat less severe.
Advances in molecular genetics have identified numerous genes responsible for SRPS including DYNC2H1 , DYNC2LI1, NEK1 , IFT140 , EVC 1, EVC2 , KIAA0586 , CEP120 , WDR19, WDR34, WDR35, WDR60, TTC21B , IFT172 , IFT80 , IFT52, IFT81, ICK, INTU, and C21ORF2 . These genes are thus candidate genes for at-risk families that can aid in prenatal diagnosis. However, cases remain that are not due to these genes, which can make molecular diagnosis challenging. However, mutations in DYNC2H1 are the most common molecular cause for the SRPS and account for about 40% of the cases. The term skeletal ciliopathies is used for these disorders because the aforementioned genes all have a profound effect on the function of the cilia.
Manifestations of Disease
Clinical Presentation
The diagnosis of SRPS is suspected by ultrasound (US) findings including short, horizontal ribs with a narrow thorax; markedly short limbs (micromelia); polydactyly; and visceral anomalies. There are subtle differences between the four types, but all are lethal. Death occurs usually within a few hours after birth because of respiratory compromise secondary to pulmonary hypoplasia or asphyxia or both.
Neonates with type I SRPS are often hydropic, with long narrow thoraces, severely shortened limbs, and postaxial polydactyly ( Fig. 55.1 ). Visceral anomalies include atrioventricular septal defects and ventral septal defects, pancreatic and renal cysts, genital anomalies, and imperforate anus. Cases with ambiguous genitalia and sex reversal have been reported.
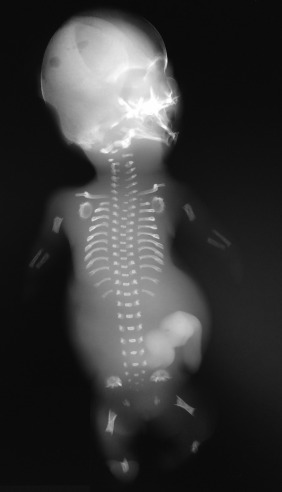
Fig. 55.1
Postdelivery radiograph of a fetus with SRPS type I. Note the micromelia, poor mineralization of the calvarium, abnormal pelvis, and spikes at the metaphyseal ends of the bones.
Type II is characterized by cleft lip, low-set ears, preaxial and postaxial polydactyly, and severe tibial shortening. These infants often have ambiguous genitalia, hypoplastic glottis, malformed larynx and tracheal stenosis, glomerular and renal tubular cysts, pachygyria, and a small cerebellar vermis. Hydrops is possible, and both ventriculomegaly and narrowing of the corpus callosum have been reported.
Type III SRPS is similar to type I SRPS but with a better mineralized appendicular skeleton. Death is usually due to asphyxia instead of pulmonary hypoplasia.
Type IV is similar to type II except that often in type IV the fetus or neonate does not have polydactyly, and the tibial shortening is less severe. Hydrops, median cleft lip, narrow chest, and bowed legs were documented in the original cases by Beemer and colleagues. Other clinical features are accessory frenulum, anophthalmia, and transverse palmar crease. Visceral findings include renal cystic dysplasia, absent internal genitalia, pancreatic cysts, intrahepatic bile duct cysts, and periportal hepatic fibrosis.
Jeune syndrome or ATD is very similar to SRPS type III, though it is less severe, and one-third of infants survive the newborn period, with many long-term survivors reported. Long-term survivors have significant medical complications that include retinal degeneration, hepatic fibrosis, polycystic liver disease, pancreatic fibrosis, and renal failure (cystic kidneys).
Imaging Technique and Findings
Ultrasound.
Skeletal anomalies detectable by US include severe micromelia often with hypoplastic/bent bones ( Fig. 55.2 ), short ribs with narrow thorax ( Fig. 55.3 ), and polydactyly ( Fig. 55.4 ). Visceral anomalies may also be noted ( Fig. 55.5 ). Three-dimensional (3D) or four-dimensional (4D) US is useful in the evaluation of fetuses with skeletal dysplasias, especially for imaging the face and extremities.