The word
sarcoma is derived from the Attic Greek word σ&aacgr;ρκωµα or
sarkoma meaning a fleshy excrescence (
1). The term was used by Galan with particular reference to a nasal growth (
2). An early English language dictionary from 1657 continued this definition of sarcoma as a “flesh growing in the nostrils like the proud flesh in a sore” (
3). In modern usage, however, soft tissue sarcomas are defined as all malignant tumors of nonepithelial, extraskeletal tissues including the peripheral and autonomic nervous system but excluding the hematopoietic system, glia, and supporting tissues of specific organs and viscera. Soft tissue sarcomas constitute approximately 6.5-7% of childhood cancer. Within this 6.5-7%, approximately one half are rhabdomyosarcoma (
4,
5,
6). Because the distribution of the types of other soft tissue sarcomas in children is distinctly different from that in adults, Spunt and Pappo have advocated the term “nonrhabdomyosarcoma soft tissue sarcoma” (NRSTS) (
7). This group of soft tissue sarcomas constitutes 3-3.5% of childhood cancers (
8), affecting approximately 500 children less than 20 years of age in the United States each year. NRSTS has not received the same focus of attention as rhabdomyosarcomas—though similar in incidence—perhaps because they have proved to be more resistant to conventional chemotherapy and radiotherapy (RT). While the surveillance, epidemiology and end results (SEER) database does not distinguish among rhabdomyosarcomas and NRSTSs, pediatric soft tissue sarcomas in the United States have been associated with improving survival over time. For patients less than 16 years of age diagnosed with a soft tissue sarcoma in 1975-1977, the adjusted 5-year survival was 61%. For those diagnosed in 1996-2004, there was a statistically significant improvement in 5-year survival at 74% (
9,
10). But to what degree this simply reflects improvements in rhabdomyosarcoma management is difficult to know. Moreover, very few patients with NRSTSs have been enrolled in cooperative group clinical trials. For instance in the United States prior to the 2006 Children’s Oncology Group (COG) ARST0332 trial, less than 200 patients had been enrolled in only three prospective clinical trials in the United States (
11,
12,
13).
For most children with NRSTS, the origin or cause of the tumor is unknown. Some cases may be traced to prior radiation exposure (see
Chapter 20), chemical exposure, iatrogenic or disease-caused immunosuppression, and neurofibromatosis, with the latter group having a 7-10% lifetime risk of developing malignant peripheral nerve sheath tumor (MPNST, historically called a neurofibrosarcoma or malignant schwannoma). The association of sarcomas with neurofibromatosis indicates that some sarcomas are associated with chromosomal deletions and translocations and the presence of abnormalities of tumor suppressor genes. Homozygous gene deletions occur in both the long and short arms of chromosome 17 in neurofibromatosis type 1. Candidate tumor suppressor genes include
17q11 (the NF tumor suppressor gene) and
p53 (17p13) (
4). Rhabdomyosarcoma and NRSTS also occur as part of the familial Li-Fraumeni syndrome. More generally in adult and pediatric soft tissue sarcomas, cytogenetic abnormalities are common with a mixture of simple translocations or point mutations giving rise to fusion genes or oncogenes and more complex genetic instability with a multiplicity of chromosome aberrations (
14,
15,
16,
17,
18,
19,
20,
21,
22,
23,
24,
25,
26,
27,
28,
29,
30,
31) (
Table 12.1). Gastrointestinal stromal tumors are of particular interest albeit rare in children since its associated
c-kit oncogene is a successful therapeutic target with imatinib, a prototype tyrosine kinase inhibitor (
32). While not yet tested in pediatric NRSTS, there are a variety of other molecular targets and associated agents under investigation in adult soft tissue sarcomas including platelet-derived growth factor receptor-A (sunitinib),
Raf kinase (sorafenib),
mTOR (rapamycin), vascular endothelial growth factor (bevacizumab), heat shock proteins,
hedgehog, histone deacetylase, and nucleotide excision repair (
31).
PATHOLOGY
The frequency of the different histologic subgroups of NRSTS of childhood varies between reporting institutions (
Table 12.2). These differences may be attributable to variations in referral patterns and to the small numbers in each series. In addition, NRSTSs often are difficult for pathologists to classify, and there is wide intraobserver variation (
33). In an M. D. Anderson Hospital series of sarcomas of the head and neck in children and adolescents, histologic diagnoses were changed in 22% of patients (
34). Several other studies have assessed discrepancy rates between the original diagnosis of soft tissue tumors and the diagnosis made by expert reviewers when patients are referred to specialty centers for entry in therapeutic trials. About 5-10% of cases having the original diagnosis of sarcoma are revised to nonsarcoma, and for 16-32% of patients with a sarcoma, the histologic subtype is revised. Where grade was analyzed, there was disagreement in up to 40% of the cases (
35). For example, malignant fibrous histiocytoma (MFH) was first described as a separate entity in the 1960s and by the 1970s was widely accepted by pathologists, identifying this entity as the most common soft tissue sarcoma subtype. Therefore during this time decades ago, the reported incidence of MFH in adults sharply increased while that of fibrosarcoma fell (
36). Historically, MFH has been unusual in childhood. But with general advances
in pathologic categorization based on immunohistochemistry, ultrastructural studies, and cytogenetics, the diagnosis of MFH has been questioned as a distinct entity (
37). At least in adults, this is no longer a common diagnosis, and when undifferentiated it is generally considered the same as a high-grade pleomorphic sarcoma.
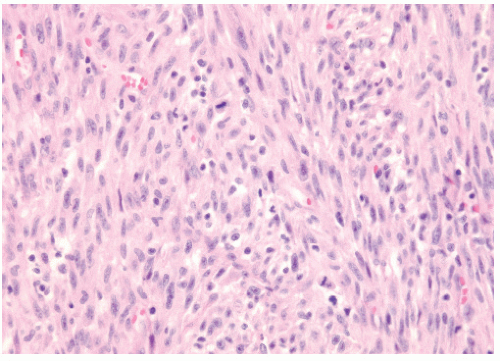
Figure 12.1 shows the histologic appearance of an MFH or pleomorphic sarcoma.
Many of the childhood NRSTSs have characteristic cell types (
Table 12.3). The WHO classification uses lines of differentiation to categorize tumors into adipocytic, fibroblastic/myofibroblastic, “so-called” fibrohistiocytic, smooth muscle, pericytic (perivascular), and vascular types, as well as tumors of uncertain differentiation (
49). Of course, other sarcomas include the skeletal muscle
tumors/rhabdomyosarcomas and bone/cartilaginous tumors discussed elsewhere in this textbook.
Malignant peripheral nerve sheath tumor (MPNST) has also been known as neurofibrosarcoma, neurogenic sarcoma, malignant schwannoma, or malignant neurilemmoma. It is a malignant neoplasm that arises in a peripheral nerve sheath. In children, from one fifth to two thirds of cases of MPNST are associated with neurofibromatosis type 1 (NF1) (
50,
51,
52). The morphology is characterized by fascicles of spindle cells with a herringbone or storiform pattern. One may observe evidence of schwannian differentiation (
53). There can be areas of significant nuclear hyperchromatism and abundant mitotic figures (
54). These tumors usually are positive for S-100, vimentin, and neuron-specific enolase. In about 2-16% of patients with NF1, nodular and plexiform neurofibromas transform to MPNSTs. NF1 is transmitted as an autosomal dominant with variable penetrance. However, almost 50% of cases are sporadic mutations (
1). People with NF1 are 1.2 times more likely to have a malignant neoplasm listed on their death certificates than those who do not have the disease. These include MPNST, optic gliomas, and pheochromocytomas (
55). NF1 is also called the von Recklinghausen disease. Friedrich Daniel von Recklinghausen (1833-1910) was born in Guterslah, Westphalia, Germany, and studied medicine at the Universities of Bonn, Würzburg, and Berlin. He was a professor in Konigsberg, Wurzburg, and Strasbourg. Also known for coining the term “hemochromatosis,” he published a monograph in 1882 that first described NF1 and the accompanying benign neurofibromas having a histologic appearance of disordered peripheral nerves and fibrosis.
Biphasic
synovial sarcoma, the more common type, also has spindle-shaped cells. These are mixed with oval and keratin-positive epithelial cells. Pseudoglandular spaces or slits and clefts mimic synovium (
56). About one third of synovial sarcomas in children are the monophasic type (
57). About two thirds of cases involve the lower extremity and one third the upper. Synovial sarcomas differ from other NRSTSs in that they have a significant risk of lymph node metastases (
57,
58). The monophasic type may have a better prognosis, but this is debatable (
57,
46). Synovial sarcomas commonly have a
t(x;18)(p 11.2;q11.2) translocation that may result in two different fusion genes
(SYT-SSX1 and
SYT-SSX2). Controversy exists as to the prognostic importance of these fusion genes. Initial reports suggested that
SYT-SSX1 fusion was associated with improved survival and less potential for metastasis (
59,
60). But in a contradictory small series, the
SYT-SSX1 fusion was associated with a 42% 5-year metastasis-free survival, compared with 89% for the
SYT-SSX2. The
SYT-SSX1 fusion is also associated with a higher cell proliferation rate as assessed by Ki-67 staining (
61). Yet another recent report suggests tumor grade rather than fusion gene subtype in synovial sarcoma is the more important prognostic factor (
62).
Liposarcomas originate from primitive mesenchymal cells rather than from mature adipose tissue. Some have a myxoid appearance, whereas others resemble benign lipomas. The pleomorphic type may resemble fibroblastic, myoblastic, or synovial sarcoma.
Angiosarcoma should always be considered high grade with an aggressive behavior, a high rate of local recurrence, a propensity to metastasize, and a poor prognosis (
63).
Leiomyosarcoma originates from smooth muscle. The well-differentiated lesions usually have a centrally located blunt-ended nucleus (“cigar-shaped”).
Fibrosarcoma is an infiltrative, fibrous neoplasm composed of interlocking bundles of spindle cells (
64,
65). The tumor usually stains positive for vimentin. There are two clinically different
forms of fibrosarcoma in children. One is a lesion appearing in the first 5 years of life with a low rate of distant spread. This type of fibrosarcoma, called the congenital type, is generally treated by excision (
4,
33,
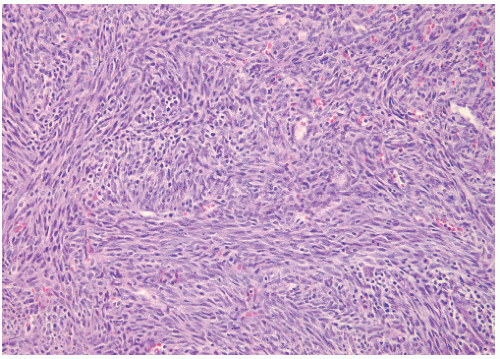
40). In children younger than 5 years of age this tumor may also be called infantile fibrosarcoma (
Figure 12.2). The other occurs in children older than 5 years and has a more ominous prognosis with behavior similar to that of adult forms (
65). This classic or adult-type fibrosarcoma is treated according to the principles outlined for other NRSTSs. Infantile fibrosarcoma has a rapid initial growth but generally indolent behavior. Local recurrence is common, metastases are rare, spontaneous regressions have been reported, and RT is rarely used. Infantile and adult fibrosarcomas are histologically identical. There is no routine microscopic, immunohistochemical, or ultrastructural way of distinguishing the two clinical types (
64).
Low-grade fibromyxoid sarcoma is an indolent tumor that rarely occurs in children. It consists of spindle and stellate cells with uniform nuclei arranged in a whorled pattern with alternating areas of fibrous and myxoid stroma (
66). True
malignant fibrous histiocytomas are pleomorphic sarcomas as noted above, often characterized by a whorled growth pattern. They are thought to arise from histiocytic cells acting as facultative fibroblasts.
Hemangiopericytomas arise from pericytes, the modified smooth muscle cell with contractile function located on the internal surface of venous capillaries and postcapillary venules (
67). The
malignant mesenchymoma has two or more cell types, any of which, taken by itself, might be considered a malignant neoplasm (
68,
69); however, the term is loosing favor as a clinicopathologic entity as such sarcomas can usually be classified in other ways (
70).
Ectomesenchymomas are felt to be of neural crest origin with multidirectional differentiation exhibiting combinations of neuroblastoma, ganglioglioma, schwannoma, embyronal rhabdomyosarcoma, benign melanocytic proliferation, and bone or cartilage elements. These rare tumors may have a dominant rhabdomyosarcomatous element with clinically similar response to chemotherapy and RT (
71) (
Figure 12.3).
There are several childhood NRSTSs that have a characteristic microscopic picture, but the cell of origin is uncertain. The
epithelioid sarcoma is a tumor of the subcutaneous tissue, tendons, and fascia, usually of the upper extremity, including the hand. There is a nodular arrangement of plump, polygonal to round, epithelioid cells interspersed with spindle-shaped cells (
5,
72). Central degeneration or necrosis is often present. The tumor tends to spread within fascial planes or aponeuroses and may grow along the neurovascular bundle and encroach on large vessels or nerves. Regional lymph node metastasis may occur in association with high-grade tumors and tumors larger than 5 cm (
73). The tumor generally stains positive for keratin.
Alveolar soft part sarcomas have a characteristic crystalline material seen with periodic acid-Schiff (PAS) stain (
74,
42). The tumor tests positive for vimentin on immunohistochemistry. The tumor cells typically have an organoid or nestlike arrangement. Vascular invasion is always seen. Of 11 children with alveolar soft part sarcoma seen at St. Jude Children’s Research Hospital in a 32-year period, 6 had localized disease and 5 had unresectable or metastatic disease. Cytogenetic studies indicate that
17q25 abnormalities are common (
42). In 19 patients seen in Italy, 4 had metastatic disease at presentation. The 5-year survival for all 19 patients was 80% (
75). A slow doubling time of the tumor may explain its very late occurrences (sometimes more than 10 years) (
37,
45).
Extraskeletal (or extraosseous) Ewing sarcoma (EOES) and
peripheral primitive neuroectodermal tumor (PNET) are characterized by cohesive, uniform, small hyperchromatic cells in a fibrous background. Dense clumping of chromatin, mitotic figures, and rosette formation are typical of PNET. On immunohistochemistry analysis, EOES are generally positive for vimentin and HBA-17. PNET is generally positive for neuron-specific enolase and other neuron-related markers such as S-100 protein, neurofilament, or HNK-1. Both PNET and EOES are associated with a particular chromosome translocation
t(11;22)(q24;q12). The progenitor cell for these two small, round, blue cell NRSTSs is not established. They may arise from neural crest, primordial germ cells, or perhaps mesenchymal stem cells (
4,
76). When PNET or EOES arises in the thoracic cavity it is called the
Askin tumor.
Desmoplastic small, round, blue cell tumor is a rare intraperitoneal malignancy occurring predominantly in adolescent boys. It is characterized by a reciprocal translocation
t(11;12)(p13;q12) associated with the
EWS-WT1 gene fusion transcript. Cells often stain positive for desmin, keratin, and neuron-specific enolase. The predominant pattern of relapse is intraperitoneal (
77). Patients generally are treated with surgical debulking, alkylator chemotherapy, and whole abdomen irradiation or intra-abdominal P-32. The relapse-free survival rate is approximately 20% (
78,
79) (
50,
51). PET scanning may be useful in follow-up for early diagnosis of recurrence (
80).
Figure 12.4 depicts a rare instance of this disease occurring outside the peritoneum.
Clear cell sarcoma is characterized by ovoid or polygonal cells with abundant clear cytoplasm, indistinct borders, large nucleoli, and abundant intracytoplasmic glycogen. Immunocytochemistry often is positive for S-100 protein, neuron-specific enolase, and melanocyte-associated antigen HMB-45. A specific chromosomal translocation t(12;22)(q13;q12) involving the DNA transcription factors ATF-1 on chromosome 12 and the EWS gene on chromosome 22 has been described in 60-75% of clear cell sarcoma cases (
81,
82).
A fair proportion of NRSTSs show no cellular differentiation. These are called undifferentiated sarcomas or sarcomas not otherwise specified.
PRESENTATION, WORKUP, AND STAGING
Most NRSTSs present as a painless swelling but may also present with signs and symptoms of vascular compression, neurologic impairment from nerve compression, or bowel dysfunction when tumors arise from the retroperitoneum.
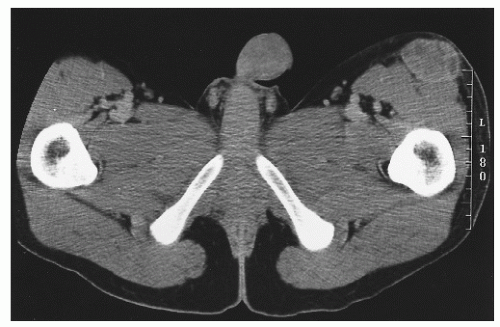
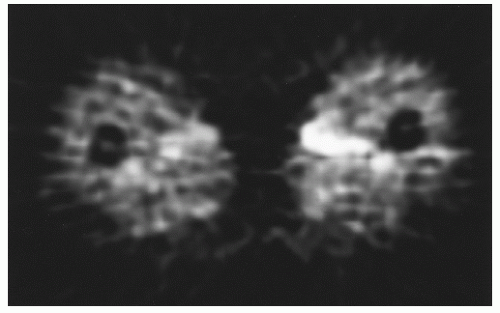
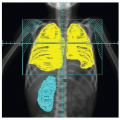
The radiographic workup begins with the plain radiograph. One looks for evidence of soft tissue mass, calcification, and destruction of adjacent bone. Radionuclide bone scanning is used to assess metastatic bone involvement, activity in bone adjacent to the tumor, and active vascular activity in the tumor itself. Arteriography is advocated for its delineation of the tumor’s blood supply, a matter of concern to the surgeon or to the direct infusional chemotherapist. Computed tomography (CT) and magnetic resonance imaging (MRI) are the essential studies for clear definition of tumor extent, patterns of infiltration, evaluation of adjacent bone, and planning surgical and radiotherapeutic approaches. MRI often shows a larger area of involvement than does CT (
Fig. 12.5). The evaluation for distant metastasis focuses on the most common site, the lungs, with chest radiograph and thoracic CT scanning (
51,
52,
56,
83,
84,
85). Metabolic scanning techniques, such as thallium scans and positron emission tomography (PET), are being used with increasing frequency. PET measures glucose utilization rate in sarcomas and may be used to assess lesion grade and to monitor neoadjuvant therapeutic response (
Fig. 12.6) (
86,
87).
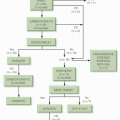
NRSTS in childhood may be staged by one of two systems. A few investigators still use the rhabdomyosarcoma grouping system, although their number is shrinking (see
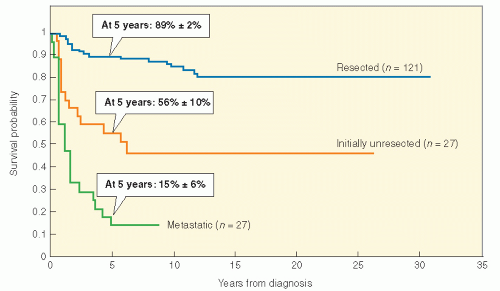
Table 11.2). This system is convenient and relies on surgical resectability as an important prognostic factor. There is no doubt that tumor size and resectability are important in predicting outcome in NRSTS of children. In fact, a retrospective analysis of NRSTS from the St. Jude Children’s Research Hospital has crudely identified three distinct risk groups: (1) grossly resected nonmetastatic disease (89% 5-year survival); (2) initially unresectable, nonmetastatic disease (56% 5-year survival); and (3) metastatic disease (15% 5-year survival) (
38,
88,
89) (
Fig. 12.7). Adverse risk factors include metastatic disease, tumor size >5 cm, high grade, positive surgical margins, intra-abdominal primary tumor site, and omission of postoperative RT in localized disease. Regarding tumor size as a prognostic factor in sarcoma patients (including rhabdomyosarcoma and NRSTS), a recent report suggests that there should be an adjustment for overall patient size (
90). The authors noted, for example, that the mortality risk associated with a patient with a body surface area of 1.75 m
2 and a 5-cm tumor was the same as for a 0.6 m
2 child with a 2.8-cm tumor.
Histologic tumor grade, however, is of considerable importance and is not directly considered in the rhabdomyosarcoma grouping system (
33,
91,
92,
93). Sarcoma grade assessment incorporates pleomorphism, spontaneous necrosis, and number of typical and atypical mitoses per 10 high-power fields. Building on the first tumor grading system (squamous cell carcinoma of the lip) in 1920, Broders and colleagues at the Mayo Clinic published a grading system for sarcomas in 1939 based on the degree of mitosis, tumor giant cells, and fibrous stroma (
94,
95). In 1969, the American Joint Commission (AJC) on Cancer Staging described a staging system for soft tissue sarcoma that uses grade, tumor size, nodal involvement, and presence of metastases as the determinants of stage (
Table 12.4). First validated by Russell et al. in 1977, the AJC system incorporates in the sarcoma staging system both TMN and tumor grade components (
96). Grade is determined by evaluation of the degree of cellularity, cellular anaplasia or pleomorphism, mitotic activity, expansive or infiltrative growth, and necrosis (
72). Frequency of distant metastases increases and the survival probability decreases with increasing size of the primary tumor (
92). About 15% of patients have metastatic disease at presentation (
4).
Histologic grading is an important way to predict the outcome of NRSTS. Moreover, as treatment decisions may often hinge on grading, the clinician should be aware of the criteria and uncertainties inherent in pathologic interpretation. There are various strategies for grading. One system, developed by the Pediatric Oncology Group (POG), labeled grade 1 as tumor that has little propensity for malignancy. Grade 2 tumors are those with fewer than 5 mitoses per 10 high-powered fields or less than 15% geographic necrosis, and grade 3 tumors are those known to be
clinically aggressive by virtue of histologic diagnosis and with more than 4 mitoses per 10 high-powered fields or more than 15% geographic necrosis (
97) (
Table 12.5). One review of this POG grading system found a 73% mortality in grade 3 lesions and 15% mortality in grade 1 and 2 tumors (
98).
The more common sarcoma grading systems that one is more likely to encounter in clinical usage are the National Cancer Institute (NCI) system and the French Federation of Cancer Centres (FNCLCC) system despite being largely based on adult cases (
99). Both are a three-tiered grading system. The aforementioned POG system is a variation of the NCI classification scheme. In the NCI system (
100), certain types of sarcomas are deemed low or high grade based on the histological subtype. A typical liposarcoma is low grade while epithelial or synovial sarcomas are always high grade, for instance. For those subtypes that are not automatically allocated to a grade, the degree of necrosis is the most important factor distinguishing intermediate versus high grade. Minor pathologic factors include the degree of mitosis, pleomorphism, cellularity, and stromal matrix. Necrosis as a prognostic marker is categorized by being minimal (0-15%), moderate (15-30%), or marked (>30%). Thus, when degree of necrosis is key to determining grade, the significant cutoff value is 15%. A grade 2 soft tissue sarcoma (e.g., a fibrosarcoma or an MFH) has up to 15% necrosis, while grade 3 disease is simply determined by greater than 15% necrosis.
The FNCLCC has similarities to the NCI system, but has a point scoring system based on tumor histology, necrosis, and mitosis that has a certain clinical appeal in its reproducibility and simplicity (
101). The main criticism, however, is that some histologic subtypes (e.g., MFH, alveolar soft parts sarcoma) do not have attributes that recapitulate normal tissues and cannot be easily scored in terms of differentiation (
Tables 12.6 and
12.7). Worldwide, the French system predominates.
Comparisons between the NCI and FNCLCC system show up to one third of cases have grading discrepancies, but the latter system may be a slightly better determinant of prognosis, albeit not for every NRSTS subtype (
99,
102). The current COG protocol on NRSTS has central pathology review; one of its aims will be to directly compare the POG and the FNCLCC grading systems for pediatric sarcomas. To date, the WHO or other pathology associations have not endorsed any particular grading schema.
While the clinician may rely on sarcoma grading to make treatment decisions, one must be aware of the degree of uncertainty in pathologic diagnosis. This problem has a bigger dimension as the pathologist is increasingly forced to evaluate smaller biopsy specimens rather than the whole tumor as newer treatment algorithms involve preoperative RT or chemotherapy. With smaller specimens, there is an increasing danger of sampling errors as necrosis, mitotic activity, differentiation, pleomorphism, and other grading aspects have geographical variability within a particular tumor. Despite advances in molecular biology, identification of cytogenetic or particular DNA or RNA lesions has not yet supplanted tumor grading to aid clinical decisions for NRSTS.