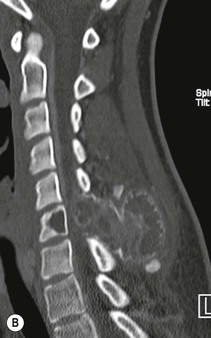
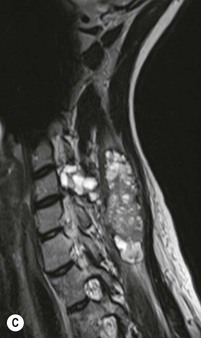
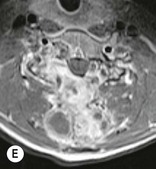
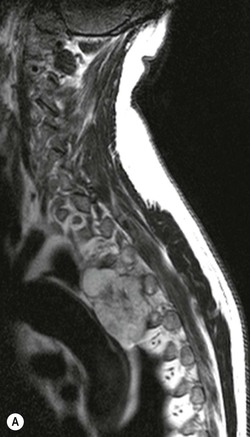
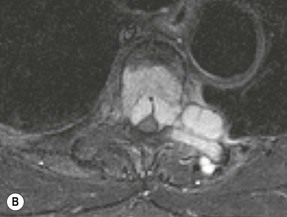
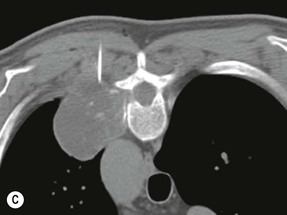
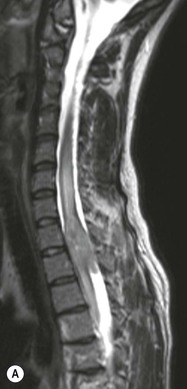
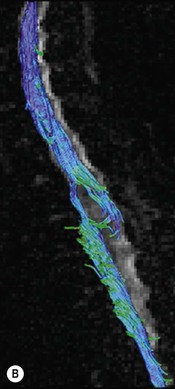
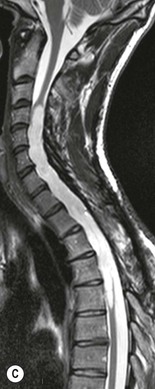
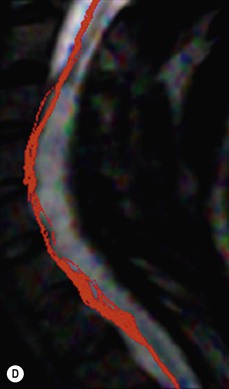
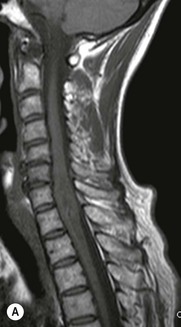
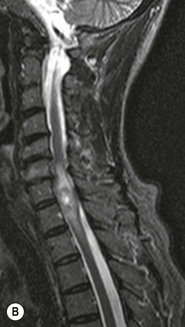
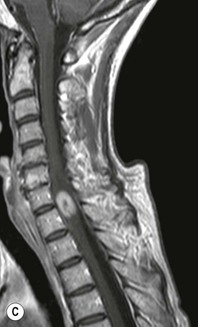
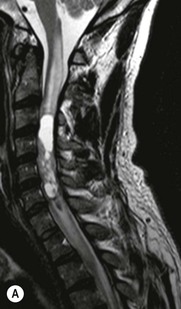
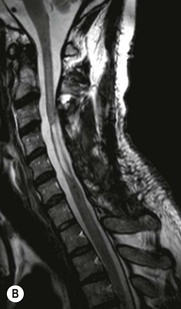
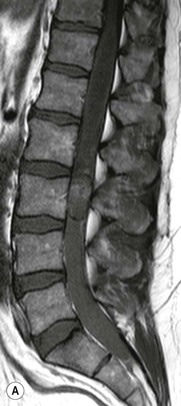
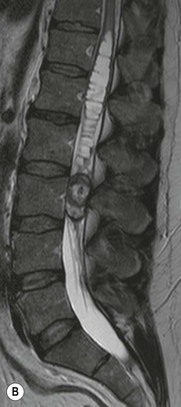
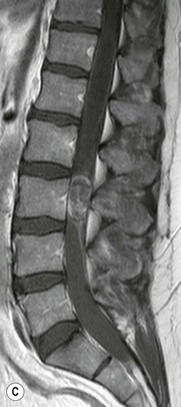
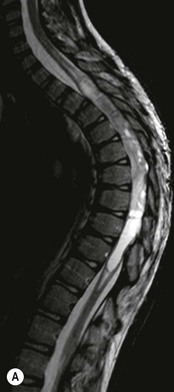
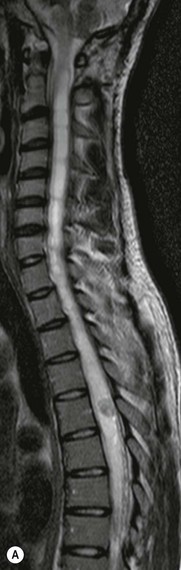
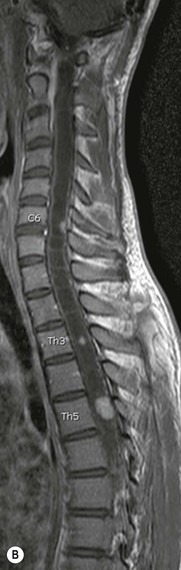
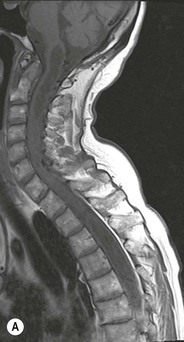
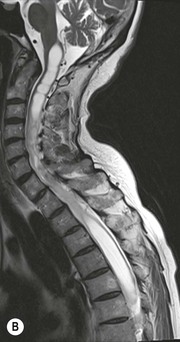
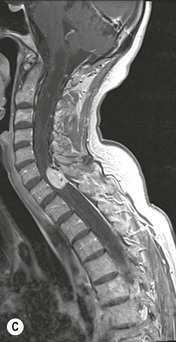
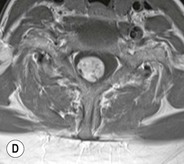
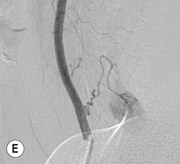
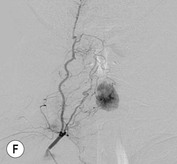
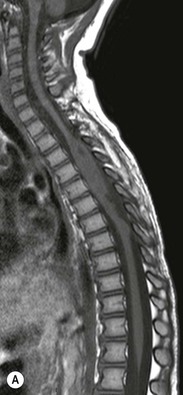
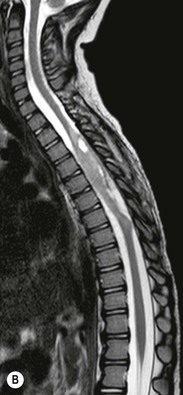
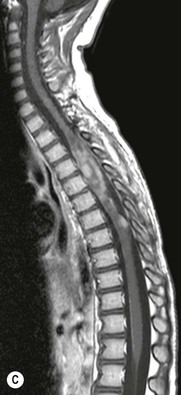
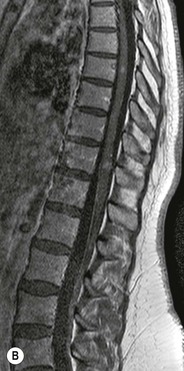
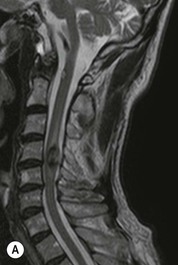
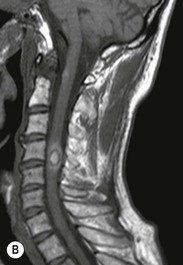
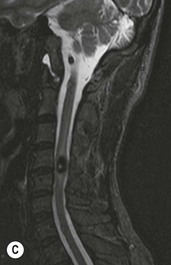
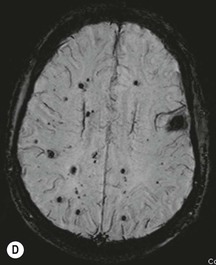
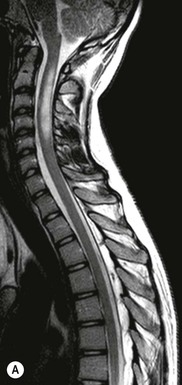
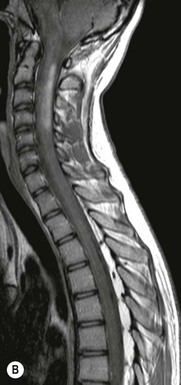
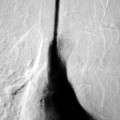
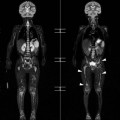
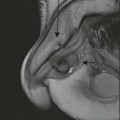
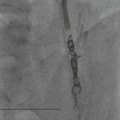
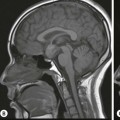
Luc van den Hauwe, Johan W. van Goethem, Danielle Balériaux, Arthur M. De Schepper † Computed tomography (CT) and magnetic resonance (MR) imaging are complementary techniques that are needed for evaluation of both the intraosseous extent of the tumour and soft-tissue involvement. MR imaging is the best imaging technique for the evaluation of the epidural space and neural structures. Plain film radiography is not the primary imaging technique of choice to image patients with spinal tumours. It is, however, often the first imaging study in the evaluation of patients presenting with back pain. Benign primary bony tumours of the spine are mostly asymptomatic and they are frequently discovered as an incidental finding when plain films are realised on the occasion of a trauma (Fig. 56-1A). Indirect findings that may be associated with intradural spinal tumours are loss of the normal cervical lordosis or torticollis in case of an intramedullary cervical tumour. Scoliosis is almost always present in cases of an extensive spinal cord tumour independent of the histology. These plain film abnormalities are more frequently encountered in children and young adults (58–81%).1 Indeed, in children, a growing spinal cord tumour (e.g. myxopapillary ependymoma) may expand the bony canal, through pressure erosion, and this enlargement of the spinal canal can be an important feature diagnosed on plain films. Also ‘scalloping’ of the posterior part of the lower thoracic and upper lumbar vertebrae may be observed in such cases. Intradural extramedullary tumours typically expand into the extradural and paravertebral space. Enlargement of the spinal foramina may be detected on plain films as well as intratumoural calcifications, when present. CT has become the optimal imaging technique for the evaluation of the vertebral bony structures. Multidetector CT (MDCT) allows for rapid and extensive visualisation of the spine and images can be reconstructed in the 3 orthogonal planes. Two-dimensional multiplanar reformatted images are useful in the evaluation of cortical bone destruction and the detection of calcifications (Fig. 56-1B) within the tumour.2 In case of intradural tumoural pathology, CT may show widening of the bony spinal canal and enlargement of the vertebral neuroforamina. Although CT and MR imaging are diagnostic methods for many cases, CT-guided biopsy may be performed for confirmation, since many bone lesions can have a similar appearance (Fig. 56-2). The imaging technique of choice for the evaluation of spinal tumours is MR imaging (Fig. 56-1C). MR imaging is also very important in the evaluation of lesions of the osseous spine.3 Protocols may vary slightly between institutions depending on the type of MR system (manufacturer, field strength, etc.), but in general several phased-array coils are used simultaneously to obtain a large field of view.4 Using advanced parallel acquisition techniques which combine the signals of several coil elements to reconstruct the image, the signal-to-noise ratio is improved along with accelerated acquisition to reduce imaging time. Parallel reconstruction algorithms that reconstruct either the global image from the images produced by each coil, or Fourier plane of the image from the frequency signals of each coil, are used to further improve image quality with better spatial resolution, and reduced artefacts.5 Full-spine and whole-body MR imaging can be performed in this manner. This is important, since it is advantageous to see the whole spine and spinal cord covered in one single examination (Fig. 56-3). Imaging protocols usually include sagittal and axial T1 and T2. T1 and T2 offer different and complementary information. T2 is superior in detecting intramedullary tumours. On the other hand, T1 is more sensitive than non-fat-suppressed T2 in detecting bone marrow disease, e.g. vertebral metastases, but short T1 inversion recovery (STIR) or other fat-suppressed T2 sequences are also able to increase the detection of certain bone marrow diseases. Suppressing the high signal of CSF in T2 sequences is very useful in detecting subtle intramedullary lesions. The most common technique to obtain this kind of image is FLAIR (fluid-attenuated inversion recovery), a sequence that nulls out CSF signal. Gradient-echo (GRE) images are useful in order to detect haemorrhagic components often present in spinal cord tumours. In screening for vertebral metastases, an additional sagittal GRE so-called out-of-phase sequence can be used. In the normal adult human, the medullary bone of the vertebral bodies contains approximately equal amounts of water and fat protons. In out-of-phase conditions, the signal of both will cancel out, leaving the vertebrae completely black. In case of vertebral pathology, however, the signal will increase and, as such, vertebral metastases (or other lesions) will clearly stand out.6 Gadolinium-enhanced sequences, usually performed in the sagittal and axial plane, are used in order to better identify solid enhancing tumour components, and to differentiate tumoural cysts whose borders enhance from associated, so-called reactive, pseudocysts.7,8 Coronal images may be helpful for the evaluation of paravertebral soft-tissue extension and fat suppression can be used to better demonstrate tumoural enhancement.2 Some authors recommend contrast-enhanced 3D-GRE T1 techniques in screening for intradural tumour dissemination.9 When dealing with extradural tumours, the administration of gadolinium is also useful for biopsy in that it allows differentiation of enhancing viable tumour from areas of non-enhancing necrosis (Fig. 56-2). Moreover, gadolinium-enhanced images better demonstrate epidural extension of the tumour.2 Diffusion-weighted imaging (DWI) and diffusion tensor imaging (DTI) have been proven useful in brain tumours. However, these techniques are much more difficult to apply in imaging of the spinal cord. Obtaining spinal cord DWI and DTI has a number of challenges. The spinal cord’s small size requires the use of small voxel sizes (higher matrix) for spatial resolution that decreases the signal-to-noise ratio. Images may be degraded because of macroscopic motion related to physiological cerebrospinal fluid pulsations, breathing and swallowing. In addition, local field inhomogeneities reducing the image resolution and the use of echo-planar sequences, typically used in brain imaging, further increases susceptibility effects.10 Although still under development, fibre tracking based on DTI holds great potential in visualising the fibres within the normal and diseased spinal cord. The effect of a growing spinal cord tumour on the fibre tracts has been demonstrated. If the lesion displaces the fibre tracts rather than infiltrating them it is is suggestive of a well-circumscribed tumour such as ependymoma (Fig. 56-4A, B), which pathologically has a plane of resection between the lesion and the normal spinal cord allowing for a surgical resection compared with a diffusely infiltrating tumour such as fibrillary astrocytoma.10–13 Bone scintigraphy can be performed when multifocal vertebral lesions with increased radionuclide uptake are suspected. However, bone scintigraphy is limited in its capacity to depict detailed surgical anatomy, particularly compared with CT or MR imaging.2 Moreover, positive findings may also be attributed to degenerative changes of the spine. Positron emission tomography (PET) has been used extensively to evaluate the grade of malignancy in brain tumours and to differentiate recurrent tumours from radiation necrosis after radiation therapy. In other regions, PET has been used to detect neoplastic lesions such as metastatic ones, and to differentiate neoplastic from non-neoplastic lesions.14 Only a few reports on the use of PET in patients with intramedullary tumours have been published. Wilmshurst et al. reported the use of both 18F-fluorodeoxyglucose (FDG) and 11C-methionine (MET) and found a correlation with histological malignancy.15 FDG-PET imaging is also useful in evaluating tumour progression and identifying the most metabolically active components in spinal cord tumours. A prospective study of larger numbers of patients with a wider range of tumour types is required, but this is difficult to achieve given the rarity of spinal cord tumours.14,15 Spinal tumours may be classified in different ways. The World Health Organisation (WHO) classification of spinal tumours is a universally accepted histological classification. The 2007 WHO classification is based on the consensus of an international Working Group of 25 pathologists and geneticists, as well as contributions from more than 70 international experts overall, and is presented as the standard for the definition of CNS tumours to the clinical oncology and cancer research communities worldwide.16 The WHO classification of CNS neoplasms is based on the assumption that the tumour type results from the abnormal growth of a specific cell type. The WHO classification also provides a grading system for tumours of each cell type and allows the classification of tumours to guide the choice of therapy and predict prognosis. Based on the grading system, most tumours are of a single defined grade. Although the updated WHO classification does not have a direct impact on the daily practice of the (neuro)radiologist or in the interpretation of images, it is valuable in the communication between clinicians, radiologists and pathologists.4,16 Based on their location on imaging findings (MR imaging, and myelography in the past), spinal tumours may be characterised as intramedullary, intradural extramedullary, and extradural spinal tumours.17 Although this classification is somewhat of an oversimplification, since lesions can reside in several compartments, this approach is very helpful as it narrows the differential diagnosis when a tumour is found in one of these anatomical compartments. Extradural lesions are the most common (60% of all spinal tumours), with the majority of lesions originating from the vertebrae. Metastatic disease is the most frequent extradural tumour, while primary bone tumours are much less frequently observed. Intradural tumours are rare, and the majority are extramedullary (30% of all spinal tumours), with meningiomas, nerve sheath tumours (schwannomas and neurofibromas) and drop metastases being the most frequent ones. Intramedullary tumours are even more uncommon lesions (10% of all spinal tumours). Astrocytomas and ependymomas comprise the majority of the intramedullary tumours.6 Primary tumours of the spinal cord are 10 to 15 times less common than primary intracranial tumours and overall represent 2 to 4% of all primary tumours of the central nervous system (CNS).17 They occur with an incidence of 1.1 cases per 100,000 persons. A considerable number of different intramedullary tumours exist; only a few of them are expected to be encountered in a routine practice. The majority of intramedullary tumours are glial tumours; about 90% are ependymomas or astrocytomas. The most frequently encountered neoplasms in adults are ependymoma (40–60%) and astrocytoma. Haemangioblastoma is the third most frequent intramedullary tumour found in adults, but it is rarely seen in children. Astrocytomas are the most common intramedullary tumour in the paediatric age group (60–90% of cases), followed by gangliogliomas. Ependymomas are uncommon in children outside the setting of neurofibromatosis type 2 (NF-2).18 Astrocytomas and ependymomas are more frequent in the thoracic and the cervical region, respectively, while myxopapillary ependymomas are typically seen in the region of the conus medullaris, filum terminale and cauda equina.5,6 MR imaging is the preoperative study of choice to narrow the differential diagnosis and guide surgical resection.19 Differentiation between ependymomas and astrocytomas before surgery is important for the surgeon because ependymomas of the spinal cord are relatively well circumscribed and they can apparently be completely removed, whereas astrocytomas have a tendency for infiltrating growth that makes complete removal difficult.20 Ependymomas are the most frequent intramedullary tumours in adults; in children these tumours occur sporadically and may be associated with NF-2. Most NF-2-related ependymomas are small intramedullary nodules that may be multiple.18 The peak incidence for spinal ependymomas is in the fourth and fifth decade, but these tumours also are found in younger patients. Ependymomas arise from the ependymal cells lining the central ependymal canal and, therefore, are frequently located centrally within the cord.7 This central location explains the more frequently observed sensory symptoms that result from the close proximity to the spinothalamic tracts.21 Motor deficits only present in the later stage of the disease, thereby delaying the diagnosis. In contrast to sporadic tumours, the majority of NF-2-related spinal tumours are asymptomatic.22 Intramedullary ependymomas are most often found in the cervical cord and less frequently also the upper thoracic cord. Most ependymomas are low grade (WHO grade 2) with a benign indolent course. The tumours are well demarcated and compress the adjacent cord rather than infiltrating it. Malignant histological subtypes (anaplastic ependymoma; WHO grade 3) rarely occur. There are four histological subtypes of CNS ependymomas: cellular, papillary, clear cell and tanycytic.6 The cellular form is the most common intramedullary variant.21 The prognosis for patients with spinal ependymoma depends on the tumour grade, degree of resection and presence or absence of CSF dissemination.21 In NF-2-patients, these tumours seldom require intervention, even for tumours that expand the cord or have associated cysts. Close surveillance with MR imaging is a reasonable option.22 CT may show canal widening, scoliosis and vertebral body scalloping. On MR imaging, ependymomas appear typically as central well-circumscribed iso- or hypointense lesions on T1 (Fig. 56-5A) and as iso- or hyperintense on T2 (Fig. 56-5B).21 Most ependymomas do enhance vividly (Fig. 56-5C) and homogeneously in 91% of the cases. They have usually well-defined borders (Figs. 56-5C, 56-6A), which allows total removal of the tumour in most cases (Fig. 56-6B).8 Because of their compressive rather than infiltrative nature, a cleavage plane may occasionally be seen on imaging. Diffusion tensor imaging may show how the tumours displace the fibre tracts rather than interrupt them (Figs. 56-4A, B). However, this may also be observed in spinal cord astrocytoma (Figs. 56-4C, D). While astrocytomas usually are very extensive, the mean tumour size of ependymomas is usually three vertebral segments. A so-called ‘cap sign’ is seen in 20–25% of cases and corresponds to low signal intensity areas seen on T2 and even better on gradient-echo T2*, capping at both sides the tumour limits (Fig. 56-6A). Those caps are haemosiderin deposits due to chronic haemorrhage. When present, the cap sign is highly suggestive for the diagnosis of ependymoma.21 Associated satellite cysts are seen in 60% of the cases, and they may be very large. Delineation of these cysts is easier after gadolinium injection. Syrinx is also a characteristic finding, especially with cervical ependymomas. Spinal cord oedema on either side of the tumour is variable, but often seen in the large multisegmental tumours.4 Myxopapillary ependymoma, a WHO grade 1 lesion, is a relatively common spinal intradural neoplasm of the conus medullaris and filum terminale arising from ependymal cells of the filum terminale. It is found predominantly in children and young adults, although it may be observed at older age. There is a slight male preponderance. Patients typical complain of chronic low back pain exacerbating during night. Myxopapillary ependymomas are slow-growing tumours, so they may become very large before the diagnosis is finally made. Associated scalloping of the vertebral body, scoliosis and enlargement of the neural foramina may be observed. Haemorrhage may occur, explaining the sudden worsening of clinical symptoms with occurrence of leg weakness and sphincter disturbances. This greater tendency for haemorrhage may also lead to subarachnoid bleeding and superficial siderosis. On MR imaging, the lesion is iso- to hyperintense on T1 (Fig. 56-7A) and hyperintense on T2 (Fig. 56-7B). The hyperintense signal may be explained by their mucin content. The tumour enhances strongly and is somewhat inhomogeneous after gadolinium injection (Fig. 56-7C). Haemorrhage and cyst formations are common features that contribute to signal inhomogeneity. The main differential diagnosis is with nerve sheath tumours, such as schwannomas.18 Although myxopapillary ependymomas are WHO grade 1 lesions, spontaneous and postoperative CSF dissemination along the craniospinal axis as well as dissemination following extradural manipulation of the tumour during spine surgery or following spinal trauma has been reported.23 In our experience, postoperative radiotherapy is therefore very useful to prevent recurrent disease. Astrocytomas are the most common intramedullary tumours (up to 90%) in children18 and account for about 30% of intramedullary tumours in adults. The peak incidence for spinal astrocytomas is in the third and fourth decade.4,6 A slight predominance among males (55%) is observed in larger series. Histology is the most important prognostic variable. In the paediatric age group, astrocytomas are mostly tumours of low grade (i.e. pilocytic and fibrillary astrocytomas).19 Pilocytic astrocytomas (WHO grade 1) account for 75% of all intramedullary tumours in the paediatric age group and typically affect children between 1 and 5 years of age, whereas fibrillary astrocytomas (WHO grade 2) account for 7% and tend to occur in older children (around 10 years of age).18 In adults, the majority (75%) are low-grade (WHO grade 2) fibrillary astrocytomas with 5-year survivorship exceeding 75%. High-grade spinal cord gliomas (WHO grades 3 and 4) are less common and associated with a poor survival. Regardless of WHO grade, spinal cord astrocytomas are infiltrative and associated with poorly characterised boundaries and, consequently, are typically biopsied only since total resection is not possible.17 The most common site of involvement is the thoracic cord (almost 70%), followed by the cervical cord. They frequently involve a large portion of the spinal cord, spanning multiple vertebral levels in length (Fig. 56-8). Involvement of the entire spinal cord (holocord presentation) is common in children but quite rare in adults.24 True ‘holocord’ tumours, however, are rare. In most cases, involvement of the whole length of the spinal cord is caused by extensive spinal cord oedema rather than by a tumour. Tumours can show areas of necrotic-cystic degeneration (60% of cases), can have a ‘cyst with mural nodule’ appearance, or can be structurally solid (about 40% of cases). The solid components are iso- to hypointense on T1 (Fig. 56-9A) and hyperintense on T2 (Fig. 56-9B), whereas necrotic-cystic components are typically hypointense on T1 and strongly hyperintense on T2. The pattern of enhancement is variable and does not define tumour margins.18 For the most part, low-grade fibrillary astrocytomas do not enhance (Fig. 56-9C), although enhancement may be observed (Fig. 56-8C). Low-grade tumours may evolve over time and become more malignant tumours. Pilocytic astrocytomas, on the other hand, do enhance intensely as they do in the brain. High-grade astrocytomas and glioblastomas tend to be more heterogeneous with necrotic-cystic areas and enhance often in a patchy mode. Intratumoural haemorrhage may be observed and is best seen on gradient-echo T2*. Associated syringomyelia may occur: the borders of those associated cavities do not enhance after contrast injection (Fig. 56-8C). Haemangioblastomas are rare benign (low grade), usually richly vascularised tumours. They represent 2–10% of all spinal tumours and are seen more commonly in adults, with a peak incidence in the fourth decade.25 Hemangioblastomas can be solitary (80%) or multiple (20%), when associated with von Hippel–Lindau syndrome (VHLs).26,27 This is an autosomal dominant disease with multiple cerebellar and/or spinal haemangioblastomas (Fig. 56-10), retinal angiomatosis, renal cell carcinoma and/or phaeochromocytoma in varying degrees.27 Spinal hemangioblastomas associated with VHLs are usually diagnosed up to 10 years earlier and are associated with less severe neurological symptoms than sporadic lesions. The incidence of spinal haemangioblastomas may be as high as 88% in patients with VHLs,28 which is much more frequent than previously reported.29 Therefore, screening spinal MR imaging should be performed in patients with VHLs. Most patients with sporadic disease have a single lesion at the cervical or thoracic level (Fig. 56-11), whereas patients with VHLs have multiple lesions at all spinal levels (Fig. 56-10). Up to one-third of patients with VHLs will develop new lesions every 2 years.28 In about 75–85% of cases these are pure intramedullary lesions, but sometimes they may be intradural extramedullary with a variable exophytic component. Pure extradural tumours are very rare.30 Preoperative evaluation of the precise tumour location is important for total resection and improving the surgical outcome.30 Haemangioblastomas have two different but rather typical presentations: either a small nodular lesion located in the subpial region and surrounded by extensive intramedullary oedema or a small nodule associated with huge and extensive intramedullary cystic components (Figs. 56-10 and 56-11). The mechanism of this peritumoural cyst formation appears to be the result of an interstitial process that starts with generation of oedema. Vascular endothelial growth factor (VEGF) acting locally in the tumour or hydrodynamic forces, or both, within abnormal tumour vasculature may drive fluid (plasma) extravasation. When these forces overcome the ability of the surrounding tissue to resorb fluid, oedema (with its associated increased interstitial pressure) and subsequent cyst formation occur.31 On T1, the solid tumour nodule is isointense to hypointense relative to the spinal cord; on T2 it is isointense to slightly hyperintense. A rich vascular network in the tumour, as well as enlarged feeding arteries and dilated draining veins (Figs. 56-11B–D), may best be seen on proton density and T2. After gadolinium injection, intense and homogeneous enhancement of the subpial nodule is seen.8 Gadolinium is especially useful in order to pick up small, multiple nodules when associated with large cystic components (Fig. 56-10B). Associated cysts may have signal intensities comparable to CSF, but sometimes a rich protein content results in a higher signal intensity on T1. Symptomatic small haemangioblastomas have relatively large associated syringes, whereas asymptomatic ones do not.32 DSA is still performed to identify the feeding arteries to the tumour (Figs. 56-11E, F) and, if possible, to perform preoperative embolisation in order to reduce the bleeding during surgery of those richly vascularised tumours. Gangliogliomas, being rare tumours in adults (1–2% of all spinal cord tumours),33 are much more frequently seen in children.21,34 They represent the second most common intramedullary tumour in the paediatric age group (15% of cases) and mostly affect children between 1 and 5 years of age, as do pilocytic astrocytomas.18 These tumours are composed of a combination of neoplastic ganglion cells and glial elements. Although they typically are low-grade tumours (WHO grades 1 and 2) with a low potential for malignant degeneration, they have a significant propensity for local recurrence, and the glial element may progress to high grade.18 Surgical resection is the treatment of choice. After resection, there is a 5-year survival rate of 89%. Their preferential location is in the cervical and upper thoracic cord and may extend to the medulla oblongata through the foramen magnum.18 Gangliogliomas may extend over more than eight vertebral segments19 and holocord involvement has been described to be more frequent than in other spinal cord tumours, probably as a result of their slow growth rate.34 Gangliogliomas are typically eccentrically located (Fig. 56-12).21 On imaging, scoliosis and remodelling are common but non-specific findings.18 Calcification may be seen on CT, but it is much less common than in gangliogliomas that occur intracranially. According to Rossi et al., calcification is probably the single most suggestive feature of gangliogliomas. In the absence of gross calcification, the MR imaging appearance of gangliogliomas is non-specific and does not allow differentiation from astrocytomas.18 Gangliogliomas have highly variable MR imaging findings. Although propensity for cyst formation has been reported to be common,19 in other series gangliogliomas were predominantly solid.18 In a large series of 27 patients with spinal cord gangliogliomas, Patel et al. described several clinical and imaging findings that are characteristic of gangliogliomas: young patient age, long tumour length, tumoural cysts (Fig. 56-12B), absence of oedema, mixed signal intensity on T1, patchy tumour enhancement (Fig. 56-12C) and cord surface enhancement.33 They speculated that the mixed signal intensity on T1 may be caused by the dual cellular population (i.e. neuronal and glial elements) and, in their opinion, this feature was somewhat unique for spinal neoplasms and was uncommonly seen in cord ependymomas or astrocytomas.33 Perifocal oedema can vary from limited or absent to extensive. Contrast enhancement can be focal or patchy, and it rarely involves the whole tumour mass;18 absence of enhancement has also been described in a minority of cases.35 Less frequent tumours include intramedullary metastasis, lymphoma, epidermoid cyst, lipoma, oligodendroglioma, intramedullary schwannoma and teratoma. Although intramedullary metastases are rare, accounting for only 5% of all intramedullary lesions, their number is growing fast due to the longer survival of many cancer patients (improved chemotherapy, etc.). They are less common than leptomeningeal metastases. The lung and breast are the most common sites of primary malignancies for intramedullary spread.4 The high sensitivity of MR imaging enables easy detection of intramedullary metastases: however, there are no specific MRI characteristics. Usually, spinal cord metastases are small, nodular, well-defined lesions, surrounded by mild to extensive oedema (Fig. 56-13A). The enhancement pattern may be either ring-like or homogeneous and intense (Fig. 56-13B).36 Recently, two peripheral enhancement features on MR imaging specific for non-CNS-origin spinal cord metastases have been described: a more intense thin rim of peripheral enhancement around an enhancing lesion (rim sign) and an ill-defined flame-shaped region of enhancement at the superior/inferior margins (flame sign).37 Melanoma metastasis has a more specific appearance, exhibiting a spontaneously hyperintense aspect on T1 linked to the presence of melanin. It may be difficult or impossible to differentiate spinal cord tumours from intramedullary non-neoplastic lesions. In one series of 212 patients undergoing surgery for intramedullary spinal cord ‘tumours’, Lee et al. reported 4% of non-neoplastic lesions.38 A variety of lesions may mimic a spinal cord tumour such as vascular cavernous malformations (cavernomas) (Fig. 56-14), tumefactive demyelinating lesions in multiple sclerosis (MS), neuromyelitis optica (NMO) (Fig. 56-15) and acute disseminating encephalomyelitis (ADEM), acute transverse myelitis (ATM), spinal cord contusion and spinal cord infarction. Also, sarcoidosis and vasculitis may mimic a spinal cord tumour. The clinical picture, laboratory findings, electrophysiological testing, and, finally, additional MR imaging of the brain may help to recognise these entities properly, in order to avoid unnecessary biopsy. Cavernous malformations (also known as cavernous angiomas, or cavernomas) represent 7–10% of all intramedullary tumours. They most commonly involve the thoracic spinal cord segments. These vascular malformations may remain clinically silent for a long period of time before an acute and rapidly progressive neurological deficit occurs due to bleeding. Before the advent of MR imaging, these lesions were extremely difficult to diagnose, especially in the spinal cord, as they usually are small and do not enlarge the spinal cord. Intramedullary cavernous malformations are usually easily recognised thanks to the typical ‘black and white’ or ‘popcorn’ appearance due to areas of mixed signal intensity on both T1 and T2 or T2* (Figs. 56-14A–C
Spinal Tumours
Radiological Investigations in Spinal Tumours
Plain Film Radiography
Computed Tomography
Magnetic Resonance Imaging
Diffusion-Weighted Imaging
Bone Scintigraphy
Positron Emission Tomography
Classification of Spinal Tumours
Intramedullary Tumours
Ependymoma
Myxopapillary Ependymoma
Astrocytoma
Haemangioblastoma
Ganglioglioma
Less Frequent Intramedullary Tumours
Metastasis.
Spinal Cord Tumour Mimics
Cavernous Malformation (Cavernoma).
Related posts:
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree