Craniosynostosis is a premature fusion of cranial sutures in infants that may lead to profound changes in craniofacial shape. These changes are a result of anatomic differences between the calvarial unit and skull base portion of the skull. Growth within the craniofacial skeleton is based on two key concepts: displacement and bone remodeling. Calvaria growth in the infant requires rapid and symmetrical displacement of each of the large bones (frontal, parietal, and occipital) of the skull along with osseous deposition along the sutures and within the bone matrix. Concomitant with these growth patterns is endocranial and ectocranial remodeling of the skull bones. Each of these patterns changes rapidly in infancy, continues into childhood, and in some cases continues even into adulthood. Following closely and symmetrically behind calvarial growth is skull base and facial growth. In the growth sequence, the anterior fossa completes its growth first, followed by the posterior and middle fossa. During this growth cycle, the skull base and face follow in form to the calvaria. When the three skull base fossae are examined individually, a unique growth pattern develops within each one. The anterior fossa relies on the growth at the sphenoid, ethmoid, and frontal bones primarily using the growth at the spheno-frontal and sphenoethmoidal sutures. Growth is rapid in this area up to about 7 years of age. The middle fossa continues its growth for an even longer period, into the teenage years, with the sphenopetrosal and petro-occipital synchondroses being most affected. The posterior fossa also continues an active growth pattern into childhood and the adolescent years. The intraoccipital synchondroses complete their growth in childhood, with the spheno-occipital synchondroses remaining active into adolescence. Concomitant to this skull base growth pattern is the growth rate of the facial skeleton. Growth within the face continues until well into the adolescent era, with a spurt that occurs during puberty. Critical angulation patterns of the maxilla are finalized in adolescence and closely follow the pattern of growth of the anterior fossa. As a result, if there is any form of premature fusion of any of the skull base sutures and synchondroses, these premature fusions can lead to significant alterations of the skull and facial alignment, resulting in a variety of different craniofacial anomalies. Head shape depends on which sutures are prematurely synostosed, the order in which they synostose, and the timing at which they synostose. Craniosynostosis may be of prenatal or perinatal onset or may occur later during infancy or childhood. The earlier synostosis occurs, the more dramatic the effect on subsequent cranial growth and development. The later synostosis occurs, the less the effect on cranial growth and development. Synostosed skulls with almost normal-shaped skulls have been observed. Therefore, skull and facial morphogenesis is a complex and multifactorial development, some of which we are only just beginning to understand.
The so-called single-suture synostoses (e.g., scaphocephaly) rarely involve the skull base and its concomitant sutures. There are two potential exceptions to this rule: plagiocephaly, which is due to premature closure of one of the coronal sutures, and trigonocephaly, where there is a premature closure of the me-topic suture.
Most craniosynostoses are congenital and manifest themselves predominantly during the period of intense cranial growth (birth through 3 years of age). Along with a small posterior fossa, cerebellar crowding occurs; Chiari malformation is a not uncommon finding associated with all these factors. Measurements of the foramen magnum typically show small aperture openings and other asymmetries. Angiographic studies have also shown significant alterations in the venous outflow patterns at the skull base, which can lead to intracranial venous hypertension. Venous hypertension is thought to be secondary to narrowing of the skull base foramina, particularly in the area of the jugular bulb. These skull base abnormalities are felt to be an important cause of hydrocephalus seen especially in syndromal craniosynostosis. Today, concepts about the pathophysiology of complex syndromic craniosynostoses have shifted from mechanical models involving tension forces of the dural structures to a molecular signalling disorder model. Mutations of the fibroblast growth factor receptors 1, 2, and 3, as well as of the TWIST and MSX2 genes, are commonly associated with the different phenotypes of this group of autosomal dominant (AD) malformations. Significant brain abnormalities have been reported in all syndromes. However, whether these abnormalities are secondary to the bone disease or primary (e.g., callosal agenesis) is still controversial. Recent evidence suggests that a white matter defect might be a primary disorder.
Radiographic Signs
Craniosynostosis is initially manifested on radiographs by straight, narrow, sharp-edged sutures with marginal sclerosis. Later, the sutures become partially or entirely bridged by bone. Cranial growth is deviated in the direction of the prematurely fused suture. On three-dimensional (3D) CT, better appreciation of the small posterior fossa, cerebellar crowding, and sometimes associated Chiari malformation is possible. Also, narrowing of the skull base foramina, particularly in the area of the jugular bulb, which may give rise to intracranial hypertension, is better evaluated on CT. Additional structural intracranial abnormalities are easily shown on CT. For the surgeon, 3D CT is an excellent tool for preoperative and postoperative assessment in patients with complex craniosynostosis.
Secondary synostoses result from decreased ICP after shunting of hydrocephalus and only rarely result in microcephaly. This type of fusion does not produce craniosynostosis.
Skull deformity may also be the result of various metabolic diseases and skeletal disorders, such as the following:
Vitamin D deficiency rickets (healed)
Vitamin D–resistant forms of rickets
Hypophosphatasia
Idiopathic hypercalcemia
Mucopolysaccharidosis (MPS)
Primary hyperthyroidism
Hypothyroidism due to hormonal overdose
Table 4.5 Types of plagiocephaly
Types of Sutures
Synostosed
Relative Frequencies
Synostotic anterior plagiocephaly
Unilateral coronal
Less common than bilateral coronal synostosis
Unilateral frontosphenoidal without coronal synostosis
Very rare
Unilateral frontozygomatic without coronal synostosis
Very rare
Synostotic posterior plagiocephaly
Unilateral lambdoid
Rare
Deformational anterior plagiocephaly
No sutures synostosed
Formerly common; now uncommon
Deformational posterior plagiocephaly
No sutures synostosed
Common
Table 4.6 Craniosynostosis
Diagnosis
Findings
Comments
Premature fusion of one frontal-parietal (coronal) suture (plagiocephaly)
Anteroposterior (AP) skull film: cranium smaller on the affected side, with a thinner calvaria, temporal bone prominence, and increased convolutional markings. Sagittal and lambdoid sutures deviate toward the affected side; crista galli and falx may also be deviated. The orbit is enlarged with an elevated superolateral rim. The lesser sphenoid wing and planum sphenoidale slope laterally upward. The petrous bone is more horizontal. Lateral view: fused suture limb is not visualized or appears as a thin, sharp-edged fissure; terminates inferiorly as a linear density in the elevated lesser sphenoid wing; middle fossa is expanded.
External appearance: Unilateral frontal flattening with apparent widening of the palpebral aperture. Even bilateral coronal synostosis can cause asymmetry if areas of the suture fuse at different times.
Premature fusion of one frontal-parietal suture and ipsi-lateral frontal-sphenoid suture
Frontal region narrow, calvaria small and elongated. Sagittal suture is narrow in young infants with sharp edges and partial ossification. Anterior fontanelle is very small. Other sutures are otherwise normal, with no signs of increased ICP. Head size is macrocephalic but sometimes normal.
Normal development. Optic nerve atrophy is very rare. No signs of increased ICP. The orbits appear quite large due to the projection effect (elongated skull); in some cases, the enlargement is real.
Premature fusion of metopic suture (trigonocephaly)
Small, keel-shaped frontal bone with hyperostosis. Metopic suture is fused. Elliptical orbits with decreased interorbital distance (hypotelorism). Coronal suture bows anteriorly toward the small anterior fontanelle. Head size normal; no signs of increased ICP.
Recent studies show cognitive impairment may be due to abnormal development of frontal lobe. Metopic suture is a normal finding in children up to 2 y of age (15% of cases); it is patent by the age of 20 y in 3% of cases (metopism).
Coronal and sagittal synostosis
Often, there is no pronounced calvarial deformity, though ICP symptoms can become very severe.
If the sutures ossify at different times, the first suture to fuse determines the calvarial configuration (see sagittal and cranial synostosis).
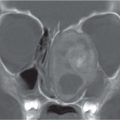
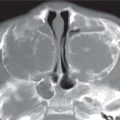
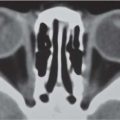
Fig. 4.11a–d Craniosynostosis of the coronal suture. (a, b) A 6-month-old girl with an abnormally shaped skull and asymmetric sphenoid wing. (c, d) 3D rendering shows a craniosynostosis of the left coronal suture.Fig. 4.12a–d Craniosynostosis. (a) A 5-month-old boy with an abnormally shaped skull. Closed right-sided coronal suture and synostosis of the right frontosphenoid suture result in a small right orbit. (b) On CT, note the flattening of the forehead on the right side. (c, d) 3D rendering shows the closed right-sided coronal suture and synostosis of the right frontosphenoid suture, resulting in a small right orbit.Fig. 4.13a, b Craniosynostosis of both lambdoid sutures. (a) Multiple dysmorphic characteristics in an 8-month-old boy with an abnormally shaped skull and scalloping of the calvaria. (b) Lateral view. Note underdevelopment of the occipital region and posterior fossa due to early closure of lambdoid sutures.Fig. 4.14a–c Dolichocephaly. (a) An 18-month-old boy with an abnormally shaped skull and scaphocephaly with frontal impressions due to craniosynostosis of sagittal suture. (b) 3D rendering shows the impressions probably due to increased ICP. (c) 3D rendering shows the synostosis of the sagittal suture.Fig. 4.15a–c Trigonocephaly. (a) A 10-month-old girl with trigonal shaped head because of an early closure of the metopic suture. (b) Note also the hypoplastic aspect of the frontal lobes. (c) Hypotelorism because of the early closure of the metopic suture.
Coronal synostosis, marked cranial deformity even in neonates. Accessory fontanelles in the frontal and parietal regions. Hypertelorism, maxillary hypoplasia; later, progressive signs of increased ICP are best visualized with CT or MRI. Absence of olfactory bulbs and tracts, midline fusion of olfactory tubercles, incomplete development of the olfactory tubercle and hippocampus and abnormal pyramidal tracts and decussation have been described. Also abnormal corpus callosum, septal defects, gyral abnormalities, seldom gray matter heterotopia, hypoplastic white matter, and megalencephaly.
Coronal synostosis is rarely unilateral. Marked bony and soft-tissue syndactyly of hands and feet. AD inheritance. Mental deficiency in Apert syndrome may be related to limbic structures’ abnormality.
Acrocephalosyndactyly II (Carpenter syndrome)
Skull as in Apert syndrome, may include craniosynostosis due to fusion of all sutures.
Extremely rare. Postaxial polydactyly, variable syndactyly. Obesitas, male hypogonadism, and mental retardation have been reported. It is autosomal recessive.
Acrocephalosyndactyly type III (Saethre-Chotzen syndrome)
Craniosynostosis may involve the coronal, lambdoid, or metopic sutures, with late closing of fontanelles and parietal foramina, thus associating hyperostosis with ossification defects. CT and/or MRI findings are described from normal to mega cisterna magna, abnormal gyral structures, and atrophy (enlarged subarachnoid spaces).
Characterized by brachycephaly with maxillary hypoplasia, prominent ear crus, and cutaneous syndactyly; shallow orbits with eyelid ptosis; other bony abnormalities are common. Children present with a flat forehead, low set hairline, and facial asymmetry with syndactyly involving the second and third fingers and the third and fourth toes, with small distal phalanges. Mental deficiency is classically uncommon. Hypotonia, seizures, and pyramidal signs have been described.
Acrocephalosyndactyly type V (Pfeiffer syndrome)
Very few imaging descriptions in the literature. In subtype I and III, ventriculomegaly and short concave clivus. Chiari type I may be present. In subtype II hydrocephalus and small posterior fossa may be seen. Subtype II is also characterized by the cloverleaf skull shape.
Subdivided into three subtypes. Subtype I is the classical, milder form with bicoronal synchondrosis, brachycephaly and flat face, hypertelorism, and mild syndactyly with broad thumbs and great toes; it is AD, with occasional mental deficiency. Subtypes II and III are sporadic and much more severe, with marked ocular proptosis and central nervous system (CNS) involvement, elbow ankylosis, and congenital heart disease. The survival rate is usually but not always poor.
All forms of craniosynostosis can occur, even fusion of all sutures with extreme signs of increased ICP. Hypoplasia or agenesis of the corpus callosum may be seen. Nonprogressive ventriculomegaly is common. In 70%, Chiari type I is present.
May occur without hydrocephalus. AD. Brachycephaly with underdevelopment of the midface, especially the maxilla. No deformities of the hands and feet. Mental deficiency is common.
Muenke craniosynostosis
Coronal synostosis, mild maxillary hypoplasia. Hypertelorism. CT and MRI findings in majority are unremarkable. Mild degree of chronic tonsillar herniation with normal looking posterior fossa has been reported.
Uncommon. AD. Mental deficiency common.
Cloverleaf skull or Kleeblattschädel
Trilobular skull with craniosynostosis. Coronal, lamb-doid, and metopic sutures may be closed with bulging of the cerebrum through an open sagittal suture or through open squamosal sutures. Synostosis of sagittal and squamosal sutures with cerebral eventration through a widely patent anterior fontanelle may also be observed.
Severity varies and different sutures may be involved.
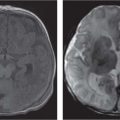
Fig. 4.16a–c Acrocephalosyndactyly type I. (a, b) CT rendering images show underdeveloped frontal calvarium and maxilla in an 18-year-old girl with Apert syndrome. (c) Note the thinning of the inner surface of the vault due to raised ICP.Fig. 4.17a, b Crouzon syndrome. (a) Crouzon syndrome in a 14-month-old boy; note the hypoplasia of the maxilla and the calvarial scalloping. (b) This intravenous contrast-enhanced CT (CECT) shows the venous collaterals because of intracranial venous obstruction.
Only gold members can continue reading. Log In or Register to continue