Definition Beckwith-Wiedemann syndrome (BWS) is a somatic overgrowth syndrome characterized by macrosomia, omphalocele, macroglossia, and ear creases.
Epidemiology Occurrence is 1 in 13,700 births (M1:F1).
Embryology BWS is caused by an imbalance of gene dosage involving a number of genes clustered at 11p15, a chromosomal region that is highly imprinted. Both genetic and epigenetic factors are implicated in the etiology of BWS. Microduplications and microdeletions on chromosome 11, as well as uniparental disomy, have been found in cases of BWS. Pregnancies conceived by assisted reproductive technologies have a higher incidence of BWS. Major malformations seen with BWS include ventral wall defects (omphalocele and umbilical hernia) and cleft palate. Congenital heart defects (20%) are common, with about 50% of the defects being cardiomegaly, which usually resolves spontaneously. Additional clinical findings include neonatal hypoglycemia, ear pits or creases, hemihypertrophy, Wilms tumor, neuroblastoma, and hepatoblastoma.
Inheritance Patterns Inheritance patterns are complex. Most cases are sporadic, but approximately 10% to 15% of families have shown autosomal dominant inheritance, with preferential maternal transmission. Careful examination of parents and genetic counseling are recommended to provide an accurate recurrence risk. Testing is available for approximately 80% of the mechanisms causing BWS, and consultation with a geneticist is recommended to discuss available testing options.
Teratogens None are known.
Prognosis Prognosis is good with successful surgical repair of omphaloceles and control of neonatal hypoglycemia. Some children require a tracheostomy, because of macroglossia, until surgical tongue reduction can be achieved. Intelligence is generally within the normal range. However, learning disabilities are increased, especially in children with 11p15 microduplications or infants with prematurity or refractory hypoglycemia. Hemihypertrophy and nephromegaly are risk factors for tumor development. The overall risk for tumor development is 8%, with most occurring by age 8.
Hemihypertrophy
Bilateral adrenal cysts
Adrenal enlargement
Prune belly
Clitoromegaly
Midface hypoplasia
Cardiac defects (25%)
Hyperplastic pancreas
Fetuses with BWS may be larger than expected by 20 weeks’ gestation.
Investigations and Consultations Required The most likely feature to be detected by ultrasonography is omphalocele. Therefore, chromosome studies, including microarray, and fetal echocardiography should be performed to exclude other causes for the omphalocele. A pediatric surgeon and neonatologist should be consulted to discuss the immediate neonatal management of the infant.
Fetal Intervention None is available.
Monitoring Third-trimester sonographic evaluations should be performed to assess fetal weight and amniotic fluid volume. To prevent the complications of extreme macrosomia, assessment of lung maturity and induction of labor may be appropriate in some situations.
Pregnancy Course The third trimester may be complicated by polyhydramnios and preterm labor. Aggressive therapy for preterm labor is appropriate to allow time for fetal lung maturity.
Pregnancy Termination Issues The subtle features of the Beckwith-Wiedemann syndrome would require an intact fetus to establish a precise diagnosis.
Delivery The significant neonatal complications in this disorder require delivery in a tertiary center. The obstetrician should be prepared for the possible complications of macrosomia, especially shoulder dystocia.
Resuscitation Most newborns with this disorder do not require intervention for the onset of breathing, although respiratory distress may be noted in association with preterm delivery as a consequence of polyhydramnios, which occurs frequently. On occasion, the macroglossia can be severe enough to compromise the airway. Oral intubation may be difficult. Use of a nasal trumpet airway is an alternative. The presence of associated major cardiothoracic anomalies may also complicate early adaptation. Management of an omphalocele, if present, consists of avoiding gastric distension and encasing the lower body in a sterile plastic bag. A bowel bag helps to prevent evaporative heat and water loss and to protect the omphalocele sac and its contents.
Transport Transfer to a tertiary center with pediatric surgery capability is necessary if there is an omphalocele or airway compromise. Transfer also is recommended to confirm the diagnosis, manage the almost-universal hypoglycemia, and establish the multidisciplinary follow-up usually required.
Testing and Confirmation Careful physical examination combined with diagnostic imaging form the primary methods of diagnosis. Molecular genetic studies, if not done prenatally, are indicated, as is careful assessment of the parents to facilitate further genetic counseling.
Nursery Management In addition to the unique management requirements for surgical repair of an omphalocele, two other major issues are management of hyperinsulinemic hypoglycemia, which can be severe and prolonged, and stabilization of the airway. Suckle feeding can be compromised by the macroglossia, in some cases necessitating alternative feeding methods. Because there is a significant association of embryonally derived neoplasia, a thorough search for such is essential. The most frequently reported site is renal, but hepatic and adrenal tumors have also been seen.
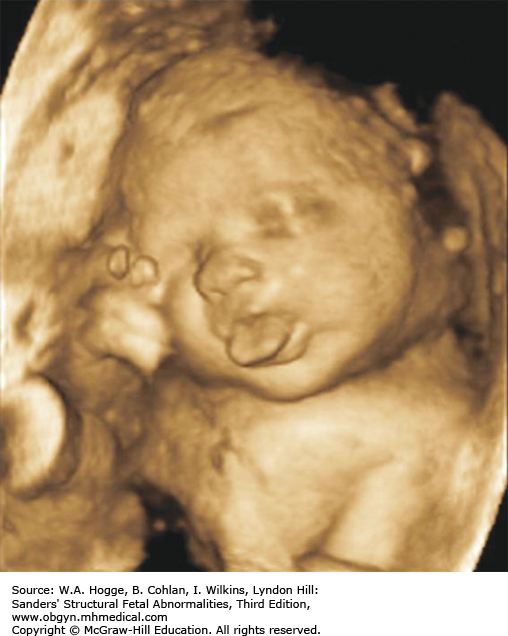
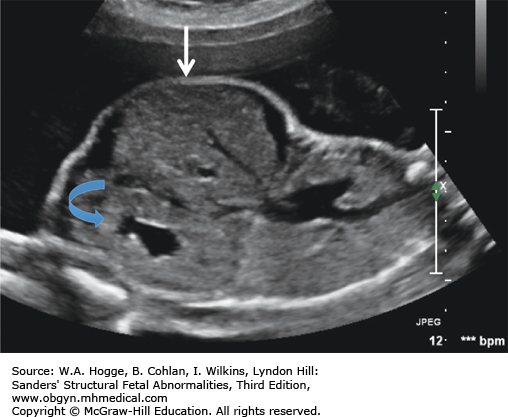
Definition Cystic fibrosis is a hereditary disorder affecting the exocrine glands. It causes the production of abnormally thick mucus, leading to the blockage of the pancreatic ducts, intestines, and bronchi and often results in respiratory infection.
Epidemiology Incidence is 1 in 3,000 newborns.
Embryology Mutation in the CFTR gene causes abnormal fluid transport across cell membranes, resulting in thick secretions. These abnormal secretions block the bronchi, resulting in recurrent respiratory infections, and the pancreatic ducts, leading to a deficiency of pancreatic enzymes. The absence of pancreatic enzymes, with abnormal intestinal secretions, leads to meconium ileus. Meconium ileus is seen in 15% to 20% of newborns with cystic fibrosis.
Inheritance Patterns Cystic fibrosis is an autosomal recessive single-gene disorder with over 1000 mutations known to be causative. A 23-mutation panel can detect 30% to 97% of cystic fibrosis carriers, depending on ethnic background.
Teratogens None are known.
Prognosis Average life span is between 30 and 40 years, with most deaths secondary to respiratory complications. Survival is higher for males than females.
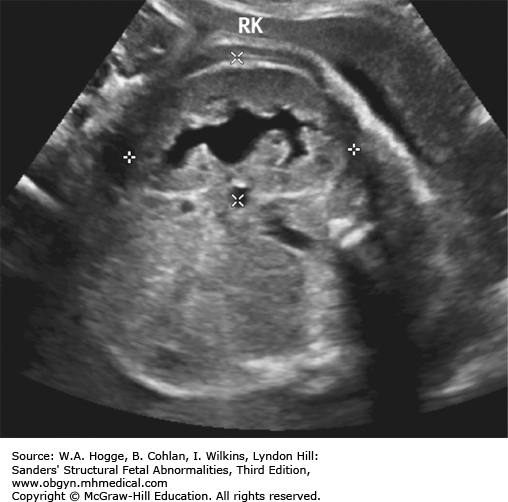
Second trimester: echogenic bowel, bright as bone
Third trimester: dilated colon with meconium
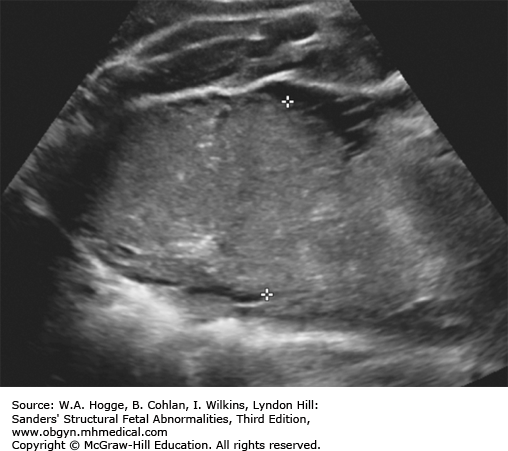
Mildly dilated loops of bowel
Dilated loops of bowel with diffuse meconium
Nonvisualization of the gallbladder
Prevalence of second-trimester echogenic bowel is 1%.
High inter- and intraobserver variability
Overdiagnosis with high-frequency (8-MHz) transducers
Risk of cystic fibrosis with second-trimester echogenic bowel is 2.5%.
A 20-fold higher risk over the general population
Risk of cystic fibrosis with the dilated loops of small bowel in the second trimester is 2.9%.
Second-trimester echogenic small bowel will detect 11% of fetuses affected with cystic fibrosis.
Nonvisualization of the fetal gallbladder has a sensitivity of 75% for detecting cystic fibrosis.
Risk of cystic fibrosis is 11.6-fold higher with nonvisualization of the gallbladder and echogenic small bowel, versus fetuses with just echogenic small bowel.
The differential diagnosis for second-trimester echogenic bowel includes
Physiologic variation of normal
Aneuploidy
Early-onset intrauterine growth restriction
Intra-amniotic hemorrhage
Congenital infection
Bowel obstruction
Investigations and Consultations Required Once the molecular diagnosis of cystic fibrosis is established, genetic counseling and referral of the family to a center with special expertise in the management of cystic fibrosis should be made. If any intra-abdominal abnormalities are noted, consultation with pediatric surgery is indicated.
Fetal Intervention None
Monitoring Periodic ultrasound examinations are indicated to assess for the development of bowel dilatation and meconium peritonitis. In the absence of intra-abdominal abnormalities, no special fetal assessment is necessary. Prenatal care should be coordinated in a tertiary center.
Pregnancy Course Polyhydramnios, secondary to intestinal obstructions, may result in preterm labor.
Pregnancy Termination Issues There are no special concerns if a molecular diagnosis has been established. Without a diagnosis, a complete autopsy and appropriate molecular studies on fetal tissue are essential.
Delivery Prenatal care and delivery should be at a tertiary center. In the absence of intra-abdominal abnormalities, routine obstetrical management is indicated.
Resuscitation No special resuscitation measures are usually required. If meconium ileus has resulted in severe abdominal distension at birth, respiration may be hindered, necessitating endotracheal intubation at birth.
Transport Delivery at a tertiary center with neonatology, pediatric surgery, genetics, and pulmonary specialists is recommended if the diagnosis is known or suspected antenatally.
Testing and Confirmation A positive state newborn screen indicates a risk of CF and needs to be followed up with genetic testing for mutations in the CFTR gene and with a sweat test. The sweat test is the diagnostic gold standard for the diagnosis of cystic fibrosis and can be performed after 2 weeks of age in term infants.
Nursery Management In cases of meconium ileus or peritonitis, transfer to a tertiary center for management is indicated (see discussion of meconium ileus/peritonitis in section 6.6). Long-term follow-up at a tertiary center with an established cystic fibrosis clinic is recommended.
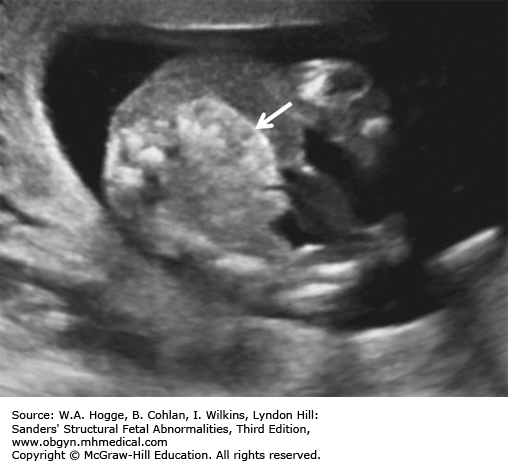
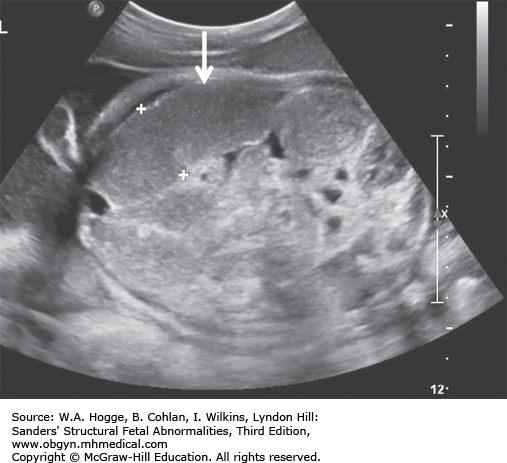
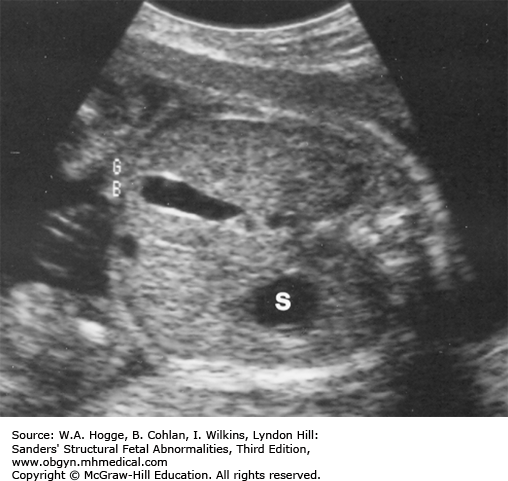
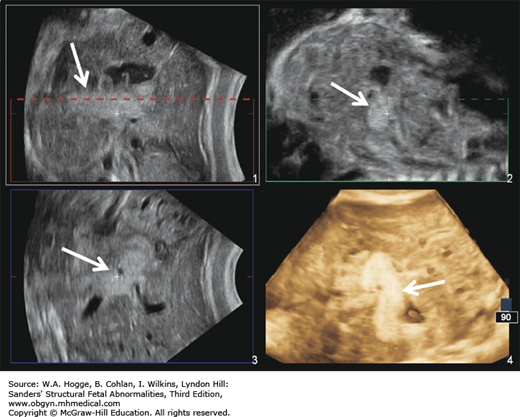
Definition Deletion of chromosome 22q11.2 is now recognized as the cause for the DiGeorge syndrome (DGS) and velocardiofacial syndrome (Shprintzen syndrome). Common features include cardiac malformations (85%), cleft palate or velopharyngeal insufficiency (48%), neonatal hypocalcemia (63%), thymic hypoplasia (65%), dysmorphic face (95%), microcephaly (40%), and moderate-to-severe learning disabilities (20%).
Epidemiology Prevalence is 1 in 4000 live births (M1:F1).
Embryology DGS represents a developmental defect of the third and fourth pharyngeal pouches, resulting in conotruncal, thymus, and parathyroid effects. Deletion 22q11.2 has been found in 50% of children with interrupted aortic arch, 35% of children with truncus arteriosus, and 16% of children with tetralogy of Fallot. It is rarely found in cases of transposition of the great vessels. It is commonly detected in children with apparently isolated cleft palate (10%) or velopharyngeal insufficiency with developmental delays.
Inheritance Patterns It is a chromosome microdeletion that is inherited in an autosomal dominant fashion, with the majority (>90%) of cases being new mutations. Because of only mild-to-moderate developmental disabilities and significant intrafamilial phenotypic variability, affected parents are found in 10% to 15% of cases. These parents are at a 50% risk of having affected offspring. Parental studies should always be performed when a 22q11.2 deletion is found in the fetus.
Teratogens None are known.
Prognosis Significant neonatal morbidity and mortality exist related to the cardiac defects and sequelae of the immunodeficiency. Extensive cardiac and cleft palate surgery may be needed, depending on associated malformations. IQ generally ranges from 70 to 90, but both speech and language development are delayed/impaired. There is significant variability in clinical and cognitive presentation. Children with 22q11.22 have a high prevalence of autism spectrum, attention deficit, and anxiety disorders, as well as psychotic-like features. Up to 30% of adolescents and adults develop schizophrenia-like psychosis.
Thymic hypoplasia (90%)
Congenital cardiac defects (80%)
Conotruncal abnormalities
Truncus arteriosus
Tetralogy of Fallot
Double-outlet right ventricle
Interrupted aortic arch
Microcephaly (40%)
Unilateral renal agenesis
Horseshoe kidney
Multicystic dysplastic kidney
Hypospadias
First-trimester thickened nuchal translucency
Intrauterine growth restriction
Polyhydramnios
The characteristic facial dysmorphology associated with DGS cannot be detected sonographically.
Severe polyhydramnios may be a harbinger of neonatal feeding difficulties.
The fetal thymus is best viewed on a three-vessel view. An absence of the thymus on this view is diagnostic for DGS.
Investigations and Consultations Required The presence of any conotruncal abnormality or cleft palate detected in utero should prompt chromosome evaluation, including a karyotype, microarray, and fluorescent in situ hybridization study for the 22q11.2 deletion. Fetal blood sampling to assess fetal T-cell function can be performed to assess immune function. Because of the significant risk of neonatal complications, consultation with a neonatologist is essential. Other consultations will depend on what other structural malformations are present. When fetal DGS is diagnosed, maternal evaluation for DGS is appropriate to eliminate subclinical phenotypic features, particularly a maternal congenital heart defect that may have an impact on labor and delivery.
Fetal Intervention This multisystem disorder is not amenable to in utero therapy.
Monitoring Polyhydramnios is a frequent complication, and both clinical and sonographic assessment of amniotic fluid volume should be done regularly. Significant polyhydramnios may result in preterm labor.
Pregnancy Course Generally, pregnancy course is unremarkable unless complicated by polyhydramnios.
Pregnancy Termination Issues If a cytogenetic diagnosis has been established, there are no special autopsy requirements.
Delivery The site of delivery must be a tertiary center where the multiple neonatal complications can be managed.
Resuscitation In general, management of the onset of respiration and the need for assistance is influenced by the nature of the associated cardiac defect. If a ductus-dependent lesion has been identified or is suspected, oxygen supplementation may be limited to 40% to 60%, while oxygen saturation is monitored to avoid hyperoxemia. Immediate infusion of prostaglandin E1 should be instituted. Other abnormalities affecting onset of respiration include orofacial clefts, Pierre Robin variants, and diaphragmatic hernia, although these are infrequent in occurrence, except with the velocardiofacial syndrome.
Transport Immediate transport to a tertiary center with pediatric cardiac capabilities, both diagnostic and surgical, is essential. During transport, the infusion of prostaglandin E1 should be maintained. Because apnea is a frequent side effect of prostaglandin infusion, intubation and assisted ventilation during transport may be necessary. Supplemental oxygen should be limited to that necessary to maintain acceptable oxygen saturation.
Testing and Confirmation The complete delineation of the cardiac defect is accomplished by echocardiography in the majority of cases. Additional laboratory testing should include serum calcium concentrations on a frequent basis, quantification of the T lymphocytes, and appropriate genetic studies, if not obtained prenatally.
Nursery Management The specific defects present will dictate the direction of care. The key issues are airway adequacy, hemodynamic stability, oxygen delivery, and the identification of concomitant problems, such as immunodeficiency and hypocalcemia. Seizures from the hypocalcemia may manifest in the absence of careful monitoring and maintenance of adequate serum calcium concentrations.
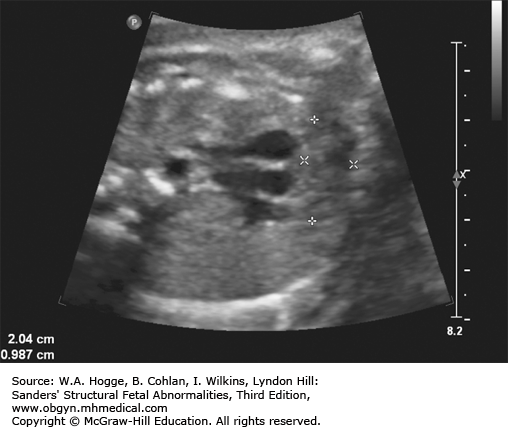
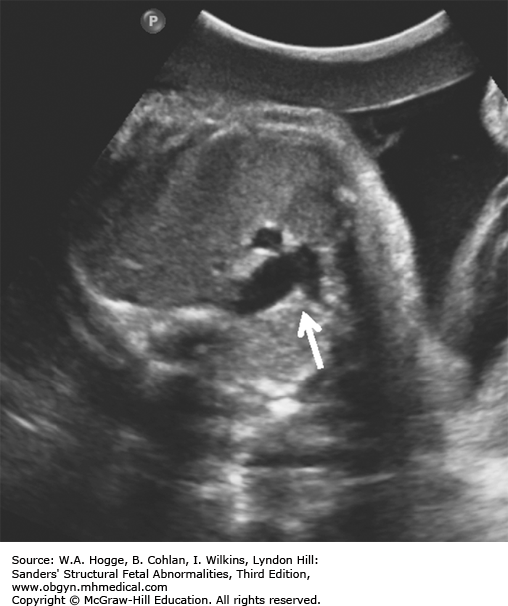
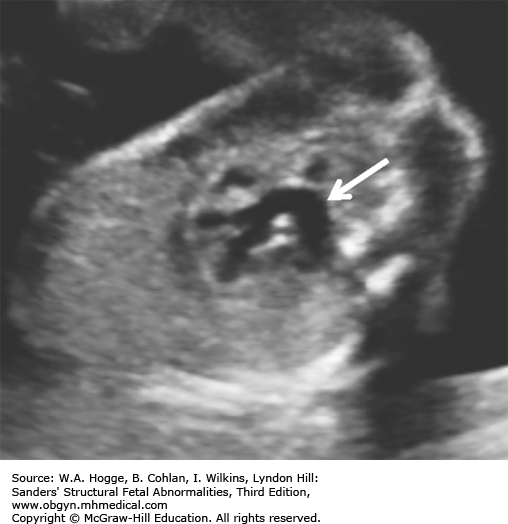
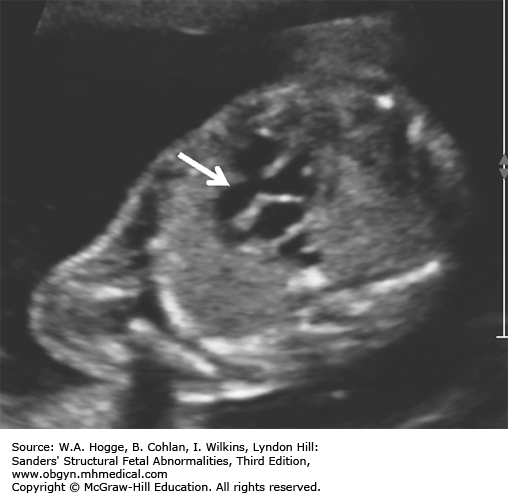
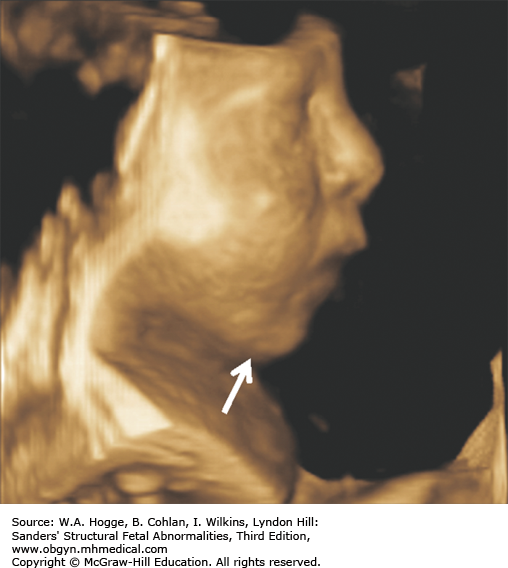
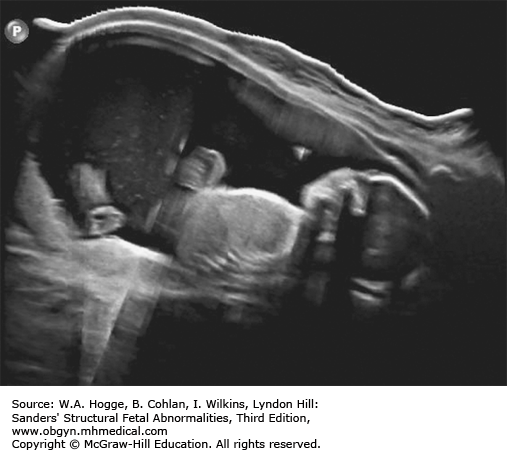
Definition Fryns syndrome is a highly lethal, autosomal recessive genetic condition characterized by congenital diaphragmatic hernia (CDH) or other defects of the diaphragm in association with other malformations.
Epidemiology Fryns syndrome is rare, with an estimated incidence of 0.7 in 10,000 births.
Embryology Diaphragmatic hernia or other defects in the diaphragm occur in 90% of cases. Other common abnormalities include the following: central nervous system malformations (Dandy-Walker malformation, agenesis of the corpus callosum); cystic kidneys; microretrognathia; genitourinary abnormalities; distal digital hypoplasia; cleft lip with or without cleft palate; and cystic hygroma. Fryns syndrome should be considered in the differential diagnosis with the detection of a CDH and any additional major congenital anomaly.
Inheritance Patterns Inheritance is autosomal recessive, with a 25% recurrence risk in future pregnancies. The gene responsible for the disorder has not yet been identified.
Teratogens None are known.
Prognosis Prognosis is poor, with most cases ending in stillbirth or early neonatal death due to pulmonary hypoplasia. In Fryns syndrome, the CDH-associated pulmonary hypoplasia is not due solely to mechanical compression. Intrinsic pulmonary arrest appears to account for a portion of CDH-associated pulmonary hypoplasia. Rare survivors without pulmonary hypoplasia had severe intellectual disability.
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


