K. Srinivasan, P. Prathiba Rajalakshmi, Sukanya Mathupal, Daspin Dhas Phakomatoses or neurocutaneous syndromes are a group of heterogeneous disorders involving the structures derived from the embryonic neuroectoderm, namely, the central nervous system (CNS), eyes and skin. The syndromes are characterized by distinctive CNS, cutaneous and oncological manifestations and usually present in childhood, with new manifestations occurring over lifetime. Each of the phakomatoses is typified by germline mutation of a tumour suppressor gene, predisposing to specific neoplastic and hamartomatous lesions. There are over 30 disease entities currently included in phakomatoses, of which the commonly encountered syndromes are enumerated in Table 2.10.1. Imaging plays a vital role in the early detection and characterization of the various lesions, identification of complications, surveillance of the affected patients and screening of family members. In this chapter, the systemic (non-CNS) manifestations of phakomatoses will be discussed. Von Hippel–Lindau disease (VHLD) is a rare inherited syndrome that predisposes to various benign and malignant tumours. It is an autosomal dominant disorder with high penetration, and more than 90% of the patients present before 65 years of age. The incidence of VHLD is between 1 in 36,000 and 45,000 live births. VHLD patients typically present in the second decade of life, with cerebellar haemangioblastoma being the most common presentation. VHLD patients have a reduced life expectancy, with an average of 59.4 years for males and 48.4 years for females. Renal cell carcinomas (RCCs) and CNS tumours are the main causes of mortality in VHLD patients. VHLD is characterized by mutations in the VHL gene, a tumour suppressor gene located on the chromosome 3p25.5. This gene encodes for pVHL protein, which plays a significant role in the regulation of hypoxia pathways through hypoxia-inducible factor (HIF). Mutations in the VHL gene cause functional loss of pVHL protein and accumulation of HIF, resulting in continuous expression of protumourigenic molecules such as transforming growth factor-alpha (TGF-α), vascular endothelial growth factor (VEGF), platelet-derived growth factor (PDGF) and erythropoietin, leading to uncontrolled angiogenesis and tumour proliferation. Inherited mutations are more common than de novo mutations, with an incidence of 80% and 20%, respectively. VHLD patients exhibit diverse clinical manifestations which correlate with the type of mutations involving the VHL gene. Based on the clinical phenotype, VHLD is broadly classified into types 1 and 2. Type 1 patients harbour truncating or missense mutations in the VHL gene. They have a low risk of developing pheochromocytoma, but high risk of developing haemangioblastoma, pancreatic cysts/neuroendocrine tumour and RCC. Type 2 patients exhibit missense mutations in the VHL gene and have a high risk of developing pheochromocytoma. Type 2 is further subdivided into 2A (high risk for pheochromocytoma, and low risk for RCC), 2B (high risk for both pheochromocytoma and RCC) and 2C (high risk for pheochromocytoma, and absent RCC). VHLD can be diagnosed clinically, if any one of the following criteria is met. VHLD displays a multitude of systemic manifestations involving the pancreas, kidneys and various other organs. The systemic manifestations of VHLD are listed in Table 2.10.2, which are elaborated in the following sections. CNS manifestations are discussed in a separate chapter. Pancreatic involvement is one of the most common manifestations of VHLD, and the reported frequency varies between 17.4% and 87.4% of patients. The commonly encountered manifestations include pancreatic cysts, serous cystadenomas and pancreatic neuroendocrine tumours (pNETs). Less common manifestations include metastases from RCC and pancreatic adenocarcinoma. Combined lesions can occur, but the association of pNET with cystic lesions is rarely reported. Pancreatic simple cysts are usually multiple and arise due to hypersecretion of VEGF. Unilocular cysts in the pancreas are uncommon in the general population, and the presence of even a single cyst in a patient with a family history of VHLD suggests a high probability of the disease. Isolated pancreatic cysts can be seen in 12%–15% of patients at the initial presentation. The cysts are usually clinically silent and are detected incidentally or during the screening of family members of VHLD patients. Rarely the cysts compress the adjacent structures including common bile duct and pancreatic duct and present with obstructive jaundice and pancreatitis, respectively. Diffuse pancreatic cysts can result in exocrine or endocrine insufficiency. The cysts appear homogeneously hypodense on CT, hypointense on T1-weighted (T1W) and hyperintense on T2-weighted (T2W) MRI sequences (Fig. 2.10.1). Cysts have thin walls with no significant contrast enhancement and may show small peripheral calcifications. Asymptomatic cysts do not require surgical intervention. Laparoscopic decompression may be necessary for patients with obstructive symptoms. Pancreatic serous cystadenoma is a benign cystic neoplasm composed of small thin-walled cysts containing serous fluid. Cystadenomas are reported in 11% of patients with VHLD. On imaging, polycystic pattern, the most common pattern of serous cystadenoma, appears as a well-circumscribed, multicystic lesion (usually more than six cysts) with lobulated contour. The size of each cyst ranges from a few millimetres to 2 cm. A characteristic feature of this lesion is the presence of a central scar, which may have a stellate calcification in up to 30% of cases. The lesion usually does not communicate with the main pancreatic duct. Less common patterns of serous cystadenomas include honeycomb pattern (numerous tiny cysts that cannot be distinguished as individual cysts) and oligocystic pattern (fewer cysts measuring > 2 cm). Patients with VHLD often have multiple cystadenomas. Rarely the lesions can be diffuse and replace the entire pancreatic parenchyma. In such cases, the differentiation of serous cystadenoma from a cluster of simple cysts is difficult (Fig. 2.10.2); however, this differentiation has no clinical significance, as both the lesions are benign and are not intervened unless symptomatic. pNET is detected in 5%–17% of VHLD patients, and the mean age of onset is 35 years. They are usually multifocal, nonfunctional and slow-growing tumours. The frequency of malignancy and metastases in these tumours is reported to be 10%–36%, whereas for sporadic pNET, the frequency is 60%–92%. Most of the lesions either present with vague symptoms or incidentally detected on surveillance. Locally aggressive tumours can present with obstructive jaundice, duodenal stenosis or gastrointestinal tract haemorrhage. Multiple pNETs are also seen in multiple endocrine neoplasia (MEN) type 1 syndrome. Islet cell hyperplasia and nesidioblastosis in the background pancreas, seen in MEN syndrome, are characteristically absent in VHLD. On CT and MRI, pNET appears as a hypervascular mass, showing intense enhancement in the arterial phase images (Fig. 2.10.3). The smaller lesions show homogeneous enhancement, and larger lesions often show heterogeneous enhancement with necrosis and calcification. The lesions are hypointense on T1W and hyperintense on T2W images. Serous cystadenoma with closely opposed cysts can be visualized as a solid-enhancing lesion and mimic pNET. Recent studies have highlighted the role of Ga-DOTATOC PET-CT as a vital screening tool for patients with VHLD. It helps in the delineation of pNET among the cystic pancreatic lesions and detects more lesions compared with CT. Libutti et al. (1998) proposed three prognostic factors that could predict metastatic disease in patients with VHLD – (1) tumour size >3 cm, (2) presence of a mutation in exon 3, (3) tumour doubling time <500 days. If none of the above criteria is met, the patient is classified as having a low risk for metastatic disease and kept on clinical and imaging surveillance, once every 2–3 years. If one of the criteria is met, clinical and imaging follow-up should be suggested once or twice in a year. If two or all the criteria are met, surgical intervention should be recommended. Pancreatic adenocarcinoma has been described rarely in patients with VHLD. The risk of adenocarcinoma in VHLD is similar to that of the general population. Pancreatic metastases from distal malignancies, particularly RCC, can be rarely seen in VHLD. They are seen as hypervascular lesions on imaging and mimic pNET. Such metastatic lesions are shown to have poor outcomes. Simple hepatic cysts can be seen in patients with VHL; however, there is no convincing evidence in the literature to suggest that there is an increased risk of developing cysts in these patients. Hepatic metastases from RCC or pNET can present as hypervascular masses in the liver parenchyma. Pheochromocytoma arises from the chromaffin cells of the adrenal medulla and is detected in 16%–30% of patients with VHLD. Extraadrenal pheochromocytomas or paragangliomas arise from the paraganglionic chromaffin cells in the organ of Zuckerkandl, carotid body, glomus jugulare, urinary bladder wall and retroperitoneum. VHLD-associated pheochromocytomas tend to present at a younger age compared with the sporadic cases, and the mean age of onset is 27 years. They are often multifocal and bilateral. The malignancy rate of these tumours in VHLD is 1%–5%, compared with 10% malignancy in sporadic cases. Multiple pheochromocytomas are also seen with MEN II syndrome and neurofibromatosis type 1. The distinguishing histological feature of VHLD from MEN-associated lesions is the absence of adrenal medullary hyperplasia. The pattern of catecholamines release from the VHL-associated tumour is continuous, in contrast to the episodic release in MEN-associated lesion. This explains the lack of paroxysmal symptoms in patients with VHLD. Most of the patients are asymptomatic, and only a few patients present with palpitations, headaches, sweating and hypertensive crisis. Diagnosis of pheochromocytoma is confirmed when the plasma metanephrine and 24-hour urine catecholamines are elevated. On CT imaging, the lesion appears as a heterogeneous mass showing necrosis with or without calcifications and typically show intense enhancement on postcontrast images. Bright hyperintense signal on T2W images and intense enhancement are characteristically seen on MRI (Fig. 2.10.4). Adrenal metastases and adenoma should be considered in the differential diagnosis of this lesion. 131I metaiodobenzylguanidine (MIBG) scintigraphy is a highly specific radionuclide imaging used for tumour localization and metastatic screening. Due to its low sensitivity, very small lesions might be missed with MIBG. PET-CT imaging using 68Ga-DOTA-TOC/NOC has more sensitivity than MIBG in diagnosing new and additional lesions, detecting metastasis and for follow-up surveillance. Adrenal cortical sparing resection of the tumour is the treatment of choice. Metachronous tumours may develop after resection. Surveillance with close follow-up imaging and biochemical markers is essential to diagnose local tumour recurrence or metachronous lesion. The renal manifestations of VHLD include cysts and renal cell carcinoma. The lesions are often multicentric and bilateral. Even if the kidneys appear grossly normal, they may harbour numerous microscopic cysts and tumours. Renal cysts are detected in 50%–75% of patients with VHLD. The spectrum of renal cystic disease comprises simple and complex cysts and cystic RCC. Patients are generally asymptomatic, even if they present with multiple cysts in both kidneys. Unlike autosomal dominant polycystic kidney disease (ADPKD), the incidence of renal insufficiency is significantly less in VHLD. Simple cysts are thin-walled and well-circumscribed lesions with no demonstrable wall enhancement on CT or MRI (Fig. 2.10.1). The simple cysts occurring in VHLD can have microscopic neoplastic cells in their walls, in contrast to those cysts seen in the general population. Complex cysts show thick and irregular walls and measurable enhancement on CT or MRI. These cysts often show an increase in enhancing areas on follow-up imaging and thus have the potential to transform into a malignant lesion. However, there is no correlation between the number and size of cysts and their malignant potential. Hence, serial imaging is recommended to document any change in morphology or increase in enhancing areas in the cysts to raise suspicion for malignant transformation. Clear cell RCC is characteristically seen in 25%–45% of patients with VHLD. They manifest at a younger age compared with the sporadic cases, and the mean age of onset is 39 years. Multifocal and bilateral tumours are common. Other hereditary syndromes that can present with renal tumours include tuberous sclerosis, Birt–Hogg–Dube syndrome, hereditary papillary renal cell carcinoma and Lynch type II syndrome. Clear cell RCC appears as a heterogeneous solid or complex cystic lesion on imaging. On CT and MRI, the solid lesions show early avid enhancement and washout on delayed images. Solid RCC shows a hypointense signal on T1W and hyperintense signal on T2W MR images. Chemical shift imaging may show a signal drop on out-of-phase images due to the presence of intracellular fat. Cystic RCCs appear as thin- or thick-walled cysts with enhancing solid components or septal nodules. MRI has superior contrast resolution than CT in characterizing the cystic masses with better demonstration of the thickening and enhancement of the walls and septae. VHLD-associated RCCs tend to be of low histological grade, slow-growing and have a better 10-year survival, compared with the sporadic RCCs. Since the incidence of metastasis from the VHLD-associated RCC is significantly less for the primary lesions smaller than 3 cm, a cutoff of 3 cm has been suggested for surgical resection of these lesions. Nephron sparing surgery (NSS) is the treatment of choice for lesions more than 3 cm in size. Radiofrequency or cryoablation are often used for treating the solid lesions lesser than 3 cm or when there are multiple lesions. Many patients develop local recurrence or de novo tumours after surgery; hence, long-term imaging surveillance is required. MRI is the ideal modality for long-term surveillance, as it avoids the radiation-related risk. Papillary cystadenoma of the epididymis (PCE) is a rare benign tumour arising from the efferent ductules of the epididymis. PCE is reported in 10%–60% of men with VHLD. PCE is usually diagnosed in young males. Bilateral tumours are pathognomonic of VHLD, whereas bilaterality is rare in sporadic cases. The patients are usually asymptomatic, and a few patients may present with a firm nodule in the region of the epididymis. On USG, the lesion usually appears as a well-circumscribed solid nodule in the head of epididymis with internal small cystic areas. Less commonly, it may appear as a cystic lesion with papillary projections. Colour Doppler shows increased vascularity within the lesion. Larger lesions may have associated ductal ectasia within the rete testis. Since these lesions have no malignant potential, they are managed conservatively and followed up with ultrasound examinations. Papillary cystadenoma of the broad ligament is a rare benign tumour reported in women with VHLD. They have a similar histological appearance to PCE with no malignant potential and are managed conservatively. Retinal haemangioblastoma is one of the common manifestations, occurring in 45%–65% of VHLD patients. The lesions develop between 10 and 30 years of age, and the mean age of onset is 25 years. Nearly 50% of the patients have bilateral lesions. Ophthalmoscopic examination shows a vascular retinal mass with a prominent feeding artery and draining vein. Small lesions may be missed on CT and MRI. When visualized, the lesions are slightly hyperintense to vitreous on T1W images and show intense enhancement on postcontrast imaging (Fig. 2.10.5). Complications include exudative or tractional retinal detachment, vitreous haemorrhage, visual deficits and blindness. Endolymphatic sac tumours (ELSTs) are benign and slow-growing lesions, often showing locally aggressive features. ELSTs arise from the endolymphatic epithelium within the vestibular aqueduct. Though they commonly occur sporadically, about 10%–15% of VHLD patients develop these tumours. Patients present with unilateral hearing loss, tinnitus and vestibular dysfunction. High-resolution CT (HRCT) of temporal bone shows an ill-defined moth-eaten or permeative destructive lesion centred at the posterior petrous bone. Intratumoural calcific foci and posterior rim calcifications are also observed on CT. MRI shows a heterogeneously hyperintense signal on both T1W and T2W images. T1W hyperintensity is due to the presence of haemorrhage, cholesterol clefts and high proteinaceous contents. Prominent flow voids may be noted in the lesions more than 2 cm in size. Heterogeneous enhancement is seen on both contrast-enhanced CT and MRI. Aggressive tumours grow laterally into the middle ear and mastoid, superiorly into middle cranial fossa, anteriorly along the petrous ridge to involve the clivus and medially into the cerebellopontine angle cistern. ELST has to be differentiated from jugular foramen paraganglioma, which is centred at the jugular bulb and rarely shows calcification. Periodic surveillance is crucial in patients diagnosed with VHLD or those with high-risk gene carriers. Surveillance detects new asymptomatic lesions, thereby increasing the median life expectancy of these patients. Various screening guidelines exist, of which the VHL Alliance surveillance is the commonly followed protocol. VHL Alliance guidelines include periodic clinical, biochemical and imaging surveillance. The various recommendations pertaining to imaging are listed in the following: Tuberous sclerosis complex (TSC) is a rare multisystem disorder, characterized by the presence of benign hamartomatous lesions in various organs. TSC has an estimated prevalence ranging from 1:6000 to 1:10,000 and has no gender predilection. TSC has a variable clinical presentation, and the classic Vogt’s triad comprising epilepsy, mental retardation and adenoma sebaceum is seen in less than 40% of affected individuals. The prognosis varies depending on the extent and severity of the manifestations. TSC is caused by mutations in either of the two genes, TSC1 and TSC2. TSC1 gene is located on the long arm of chromosome 9 (9q34) and TSC2 on the short arm of chromosome 16 (16p13). These tumour suppressor genes code for the proteins hamartin and tuberin, respectively. Both the proteins are involved in the mammalian target of rapamycin (mTOR) pathway, which regulates the cell growth, size and proliferation. Hence, mutations in these genes result in tissue overgrowth and formation of tumours throughout the body. Sporadic mutations account for nearly 70% of the mutations detected by genetic testing, and they more commonly involve the TSC2 gene. The remaining 30% of mutations are inherited in an autosomal dominant pattern with an equal frequency of TSC1 and TSC2 involvement. No mutations are detected in 15%–20% of cases with TSC using the currently available genetic tests. The proximity of the TSC2 with PKD1 gene explains the presence of multiple renal cysts in TSC. The diagnostic criteria of TSC were established by the International Tuberous Sclerosis Complex Consensus group and were updated in 2012. The identification of pathogenic mutation of TSC1 or TSC2 gene is sufficient to make a diagnosis of TSC. When no mutation could be identified in patients by conventional genetic testing, the clinical criteria could be used for diagnosis. The clinical criteria have 11 major and 6 minor diagnostic features. Based on the presence of major and minor features, a patient is assigned any one of the two categories – definite TSC or possible TSC (Table 2.10.3). TSC is usually diagnosed in early childhood, and common clinical presentations are seizures, developmental delay and hypomelanotic macules. The CNS, kidneys and skin are frequently affected in TSC. The intracranial manifestations are discussed in a separate chapter. The three characteristic manifestations of TSC, namely angiomyolipomas (AMLs), lymphangioleiomyomatosis (LAM) and clear cell sugar tumour (CCST) of lung are categorized under perivascular epithelioid cell tumours (PEComas). In 2013, the World Health Organization (WHO) defined PEComas as mesenchymal tumours composed of histologically and immunohistochemically distinctive perivascular epithelioid cells. The cutaneous manifestations may be noticed at birth or later in life. Three or more white hypopigmented macules at birth are noted in 90%–100% of cases, which are thumbprint or ash-leaf in shape. Multiple hypopigmented guttate/confetti macules are also noted. Café au lait macules (in ~30% of patients), classical of neurofibromatosis, are also manifested in TSC. Café au lait macules develop a few weeks from birth and are less than 6 in number. Facial angiofibromas (∼80%) develop between 2 and 5 years of age. These are bilateral, symmetrical erythematous papules or papulonodules in cheeks and forehead, which might further develop into the fibrous plaque of forehead (Fig. 2.10.6). Shagreen patches (collagen plaques) in the lumbosacral region are seen in half of the cases by 2 years of age. Koenen’s tumour or periungual fibroma develops during childhood, especially around toenails and manifest by early adolescence. Molluscum pendulosum, gingivitis fibroma and dental enamel pits complete the orocutaneous spectrum of TSC. Retinal hamartoma is a benign astrocytic proliferation, seen in approximately 50% of patients with TSC. The presence of multiple retinal hamartomas is one of the major features in the TSC diagnostic criteria. They usually develop over the first few months or years of life. On CT and MRI, hamartomas appear as a sheet-like or a nodular lesion arising from the retina. Moderate enhancement is usually seen on contrast imaging. The hamartomas may calcify, and in such cases, distinguishing from retinoblastoma is difficult. Punched-out areas of chorioretinal hypopigmentation or achromatic retinal patch is a minor diagnostic criterion and seen in about 5% of patients. Nonretinal findings observed in TSC include angiofibromas of the eyelids, coloboma of the lens, iris and choroid, strabismus and papilledema. Renal manifestations are seen in 48%–80% of TSC patients and comprise AML, renal cysts and RCC. TSC2 mutations result in more common and severe renal involvement. The renal lesions progress over time with new lesions detected on follow-up imaging. Renal failure is one of the leading causes of death in TSC patients. AML is the most common renal manifestation, occurring in about 80% of patients with TSC. Conversely, only 20% of patients with AML have tuberous sclerosis, while the remaining 80% occur sporadically. The presence of two or more AMLs is a major criterion in the diagnosis of TSC. Renal AMLs manifest in early childhood, and the mean age of onset is 7.2–9.2 years of life. Most AMLs are asymptomatic; however, they can present with abdominal pain, hematuria, haemorrhage or a palpable mass. Unlike sporadic AMLs, TSC-associated AMLs are usually larger, multiple, bilateral with high growth rate and more prone to haemorrhage. AML is a triphasic mesenchymal neoplasm composed of dysmorphic blood vessels, fat and smooth muscles in varying proportions. AMLs are broadly classified into classic AML and fat poor AML. Classic AMLs show areas of macroscopic fat (HU values < −10) within the lesion and account for about 70%–80% of all TSC-associated AMLs. The lesions typically appear hyperechoic to the renal parenchyma on USG. CT imaging shows a varying amount of macroscopic fat and soft tissue within the tumour. MRI confirms the presence of intralesional fat on frequency-selective fat suppression sequence. Chemical shift (CSI) MR sequence shows an Indian ink artefact at the fat–water interface between the AML and the normal renal parenchyma on opposed phase images. Fat poor AMLs affect up to 20%–30% of patients with TSC. The lesions do not possess macroscopic fat and often mimic RCC on imaging. Since RCC also occurs in patients with TSC, recognition of the fat poor AMLs is essential as both have different clinical course and prognosis. Two different radiological classifications were proposed for characterizing AMLs. Jinzaki et al. (2014) classified renal AML based on the clinical, imaging and histopathological features into classic AML, fat poor AML with three subtypes and epitheloid AML. Song et al. (2016) proposed a classification based on the CT and MRI findings into fat rich, fat poor and fat invisible AMLs. The imaging characteristics of each type are detailed in Table 2.10.4. The classic and fat poor AMLs are benign tumours, whereas epitheloid AML is a potentially malignant lesion. Percutaneous biopsy or close follow-up imaging is indicated when CT and MRI could not distinguish between the benign and malignant AMLs or when there is a diagnostic confusion with RCC. Epithelioid AML is a rare variant composed predominantly of epithelioid cells with lesser amount of fat and vessels. They usually present as large heterogeneous masses with a mean size of 7 cm. The disease course may range from entirely benign to locally invasive tumour with renal vein or IVC extension and metastatic disease. They also have high propensity to bleed. Since AML contains dysmorphic vessels with absent internal elastic lamina, they are prone to aneurysm formation and rupture. The risk of haemorrhage is high when the size of the lesion is more than 4 cm, and the size of the aneurysm is more than 5 mm. Acute intralesional haemorrhage may mask the fatty component, and the lesions are often misinterpreted as RCC. Ruptured AML is suspected when there is intrarenal, perinephric or retroperitoneal haemorrhage (Fig. 2.10.7). Massive retroperitoneal haemorrhage may be seen in up to 10% of patients and is a significant cause of mortality in TSC. TSC-associated AMLs grow at a rate of 1.25 cm/year, compared with 0.19 cm/year in sporadic AMLs. Various protocols have been proposed for the surveillance of untreated patients. A baseline USG or MRI is required at the time of diagnosis of TSC. If an AML < 3 cm is detected, follow-up imaging is indicated every 2–3 years. If an AML ≥ 3 cm is detected, CT or MR angiogram study has to be performed to look for aneurysms within the lesion. Asymptomatic patients with ≥ 3 cm lesion should be followed up annually. Patients presenting with complications of AML such as low back pain, peritoneal irritation, hematuria or hypovolemic shock should be treated immediately. Angiographic embolization of the intralesional aneurysm is indicated to control acute haemorrhage. Surgery is usually done for AML of more than 4 cm and aneurysm size of more than 5 mm. Surgical resection is the treatment of choice for the epithelioid variant. The mTOR inhibitor, everolimus, is currently approved for the treatment of adult TSC-associated AMLs, which are at risk of complications but do not warrant immediate surgery. Renal cysts are detected in approximately 18%–53% of TSC patients and are usually seen from childhood. The cysts are characteristically multiple and bilateral and show an increase in size and number over time. In nearly 2% of TSC patients, there may be associated mutations in the PKD1 gene on chromosome 16. This subset of patients exhibits early onset of renal involvement resembling polycystic kidney disease with development of hypertension and renal failure. RCC is a rare manifestation of TSC with an estimated incidence similar to that of the general population; however, TSC-associated RCCs tend to occur in younger patients and the mean age of onset is 28 years. They have a slower growth rate compared with the sporadic cases. Similar to the general population, clear cell carcinoma is the commonly reported subtype in TSC, with occasional reports of papillary and chromophobe subtypes in the literature. LAM is the most common pulmonary manifestation, occurring in 26%–39% of patients with TSC. TSC-associated LAM occurs predominantly in young females, and the average age of onset is 33 years. Pathologically, this disorder is characterized by smooth muscle proliferation in the pulmonary interstitium accompanied by cystic changes in the lung parenchyma. Exertional dyspnea, recurrent pneumothoraces and chylous pleural effusion are the main clinical manifestations of LAM. It is a slowly progressive disorder, leading to respiratory failure and eventually requiring lung transplantation. Characteristic findings on chest CT include multiple, well-circumscribed air-containing thin-walled cysts distributed uniformly throughout the lungs (Fig. 2.10.8). The size of the cysts decreases on expiratory CT images, indicating the possibility of a communication between the cysts and the airway. LAM should be differentiated from pulmonary LCH, which shows irregular shaped thick-walled cysts and is often accompanied by small nodules. LCH characteristically involves the upper and middle zones, whereas the cysts in LAM show uniform distribution. Less common differential diagnosis for pulmonary cysts includes lymphocytic interstitial pneumonia, Sjogren’s syndrome, Birt–Hogg–Dube syndrome, light chain deposition disease, amyloidosis and metastatic endometrial stromal cell sarcoma.
2.10: Systemic manifestations of phakomatoses
Von Hippel–Lindau disease
Introduction
Molecular genetics
Genotype–phenotype correlation
Diagnostic criteria
Systemic manifestations of Von Hippel–Lindau disease
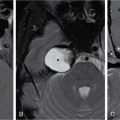
Pancreatic manifestations
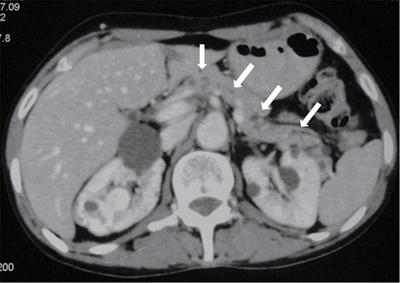
Pancreatic neuroendocrine tumours
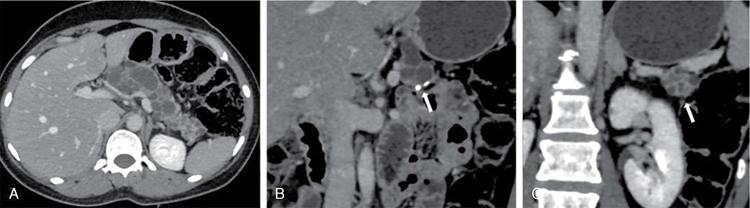
Rare pancreatic lesions
Hepatic manifestations
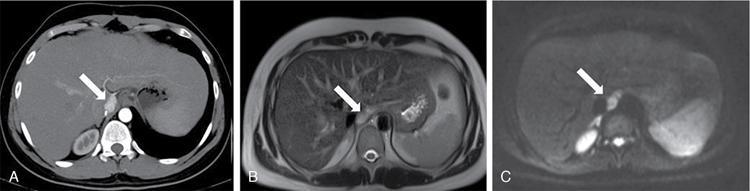
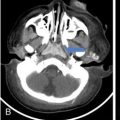
Pheochromocytoma
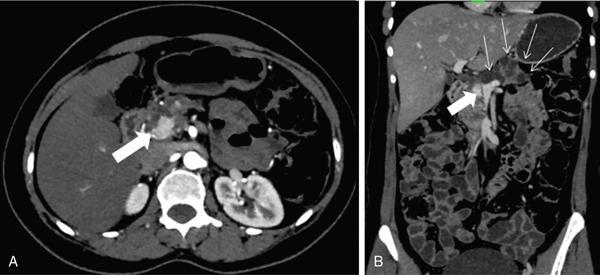
Renal manifestations
Renal cysts
Renal cell carcinoma
Papillary cystadenoma of the epididymis
Retinal haemangioblastomas
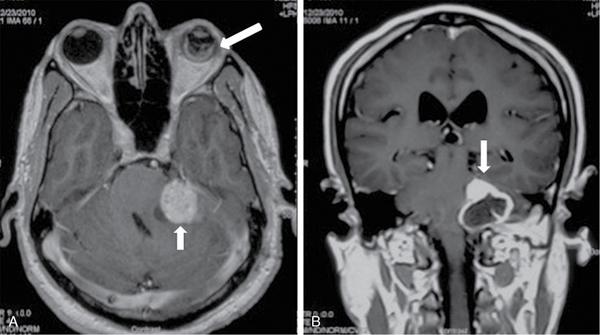
Endolymphatic sac tumour
Surveillance protocol for Von Hippel–Lindau disease
Tuberous sclerosis complex
Introduction
Genetics
Diagnostic criteria
Systemic manifestations
Cutaneous markers
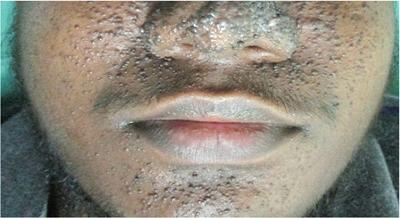
Ophthalmic manifestations
Renal manifestations
Renal angiomyolipomas
THE CLASSIFICATION PROPOSED BY JINZAKI et al. (2014)
Type of AML
Subtype
Amount of Fat
CT
MRI
Classic AML
Abundant
Macroscopic fat with attenuation value < −10 HU on plain CT
Signal loss on FS sequences and Indian ink artefact on CSI
Fat poor AML
Isoattenuating
Scattered
Attenuation values between −10 and 45 HU
Hypointense on T2W, scattered areas of fat suppression on CSI
Hyperattenuating
Few or none
Hyperattenuating (>45 HU) and homogeneously enhancing
Hypointense on T2W, no fat suppression on FS and CSI
AML with epithelial cells (AMLEC)
Few or none
Hyperattenuating (>45 HU) and homogeneously enhancing with cystic areas or multilocular cystic
Hypointense on T2W, no fat suppression on FS and CSI with cystic areas or multilocular cystic
Epithelioid AML
Few or none
Hyperattenuating (>45 HU) and heterogeneously enhancing or multilocular cystic
Hypointense on T2W, heterogeneously enhancing or multilocular cystic
THE CLASSIFICATION PROPOSED BY SONG et al. (2016)
Type
Ultrasound
CT
MRI Tumour–Spleen Ratio
Signal Intensity Index
Fat rich
Hyperechoic
Attenuation value ≤ −10 HU on plain CT
<0.71
More than 16.5%
Fat poor
Intermediate
Attenuation value > −10 HU on plain CT
<0.71
More than 16.5%
Fat invisible
Isoechoic
Attenuation value > −10 HU on plain CT
0.71 or greater
16.5% or less
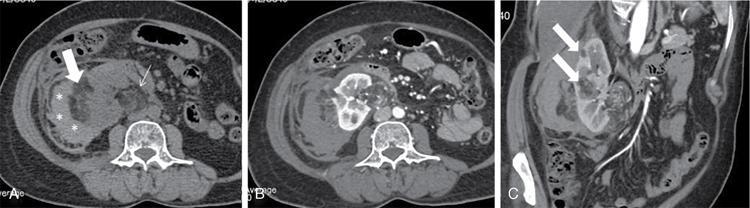
Renal cysts
Renal cell carcinomas
Pulmonary manifestations
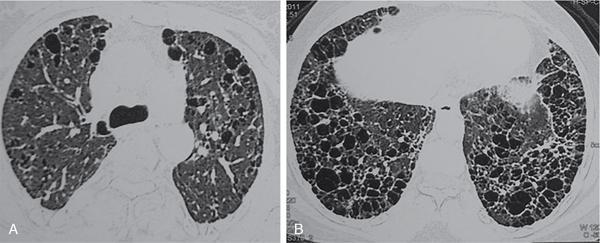
Lymphangioleiomyomatosis.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


