- •
Identification of malalignment of the conal septum anteriorly into the right ventricular outflow tract
- •
Pulmonary annulus size either equal to or smaller than the aortic annulus
- •
Large ventricular septal defect, beneath the aorta
- •
Large aorta overriding ventricular septal defect and over both ventricles
- •
Obstruction of the right ventricular outflow tract (subpulmonic, pulmonary valve, or supravalvar pulmonary stenosis)
- •
Continuity and confluence of the branch pulmonary arteries
- •
Size of the branch pulmonary arteries
- •
Identification of the ductus arteriosus, its location, origin, and connection to the branch pulmonary arteries
- •
Determine the direction of shunting across the ductus arteriosus
- •
Search for any additional muscular ventricular septal defects besides the large malalignment type of ventricular septal defect
- •
Aortic arch sidedness (left or right)
Anatomy and Anatomical Associations
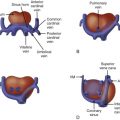
Tetralogy of Fallot (TOF) is an abnormality of conotruncal formation. The conotruncus consists of the muscularized conus or conal septum—muscular septal tissue separating the region just beneath the great vessels—and the superiorly adjacent truncus arteriosus, which eventually differentiates into the great vessels arising from the heart, the pulmonary artery and aorta. Early in cardiac development, the conotruncus originates from the primitive right ventricle but then shifts leftward over a forming ventricular septum. To complete normal development, septation and rotation of the conotruncus occurs, resulting in a right ventricle–to–pulmonary artery relationship and a left ventricle–to–aorta relationship. In TOF, there is anterior and rightward deviation of the conal septum as well as incomplete rotation of the great arteries and the aorta is much more anteriorly positioned than normal. The ensuing anatomical configuration consists of a large anterior malalignment type ventricular septal defect (VSD), an aorta that “overrides” the VSD, varying degrees of right ventricular outflow tract (RVOT) obstruction at both the pulmonary valve and the subvalvar levels, and right ventricular hypertrophy ( Figure 10-1 ). The pathological associations of TOF were first described by Neils Stenson in 1672, but it was Dr. Etienne Fallot who is credited for correlating the clinical presentation with the anatomy in 1888 referring to the condition as “la maladie bleue. ” In actuality, the term tetralogy of Fallot was not popularized until the early 1900s.

A number of anatomical variations exist in TOF. The pulmonary valve and subpulmonic region may be only modestly hypoplastic and represent mild degrees of pulmonary stenosis. In contrast, there may be severe valvar hypoplasia or even complete atresia of the pulmonary valve and main pulmonary artery segment. In pulmonary atresia, there is no antegrade flow across the pulmonary valve. The branch pulmonary arteries may originate as a confluence from the ductus arteriosus (DA) or blood flow to the lungs may be supplied by collateral vessels arising from the aorta known as major aortopulmonary collateral arteries. Moreover, the branch pulmonary arteries may have varying degrees of hypoplasia, which can affect long-term lung function. Another variant, TOF with absent pulmonary valve syndrome, occurs when the pulmonary valve leaflets are severely underdeveloped or rudimentary, resulting in severe pulmonary insufficiency and dilation of the pulmonary arteries. Associated anatomical considerations include a right aortic arch, which is present in up to 25% of patients with TOF. The DA may be tortuous or absent. Variations of coronary artery anatomy can be seen. An aberrant left anterior descending artery originating from the right coronary artery and crossing the RVOT is noted in approximately 1% to 2% of cases and has important surgical implications. Developmentally, the coronary arteries arise from the region of the ventricle and migrate toward the aorta. In TOF, the aorta is rotated and located more anterior than normal; hence, the left coronary system is prone to link up with the right, which is positioned much more leftward than normal, leading to this finding. A persistent left superior vena cava has been reported in 11% of cases. Additional muscular VSDs can occur, and the combination of TOF and atrioventricular canal defect is strongly associated with trisomy 21.
Frequency, Genetics, and Development
TOF accounts for 7% of all congenital heart disease and is one of the most common forms of cyanotic congenital heart disease, occurring in 0.3 to 0.4 per 1000 live births. Nearly one third of cases of TOF have an associated extracardiac anomaly or chromosomal derangement. Microdeletion of chromosome 22q11 (22q11 del) is present in approximately 90% of patients with DiGeorge syndrome, a genetic disorder associated with multiple extracardiac abnormalities such as palatal abnormalities, feeding and speech difficulties, thymic hypoplasia leading to immunodeficiency and hypocalcemia, varying degrees of developmental, cognitive, and psychological deficits, facial dysmorphism, as well as renal and skeletal abnormalities. Congenital heart disease is estimated to occur in 75% to 80% of patients with 22q11 del, and the most common cardiac defects noted are abnormalities of conotruncal formation, including TOF, interrupted aortic arch, and truncus arteriosus. Conversely, 22q11 has been identified in 16% to 18% of all cases of TOF, and in one half of those patients with TOF and an associated right aortic arch. Other important genetic associations with TOF include Alagille syndrome, VACTERL (vertebral abnormalities, anal atresia, cardiac abnormalities, tracheoesophageal fistula and/or esophageal atresia, renal agenesis and dysplasia, and limb defects) association, CHARGE (coloboma, heart disease, atresia choanae, retarded growth and retarded development and/or central nervous system anomalies, genital hypoplasia, and ear anomalies and/or deafness) association, and cat eye syndrome, as well as trisomy 13, 18, and 21 (especially in TOF with common atrioventricular canal defect).
Our understanding of the genetic and molecular basis of this congenital anomaly has increased considerably, although it is still far from complete. Several studies have focused on the role of transcription factors as specific regulators of steps in cardiac development. Gene mutations have been linked to the development of TOF, including NKX2-5, Tbx1 (linked in DiGeorge syndrome), as well as NOTCH 1 and 2 , and JAG1 (linked to Alagille syndrome).
Fetal Physiology
The fetus with TOF is typically quite stable from the cardiovascular standpoint. The structural anomalies of (1) an obstruction across the right ventricular outflow to the pulmonary artery and (2) a large communication (VSD) between the two ventricles lead to an alteration in fetal blood flow patterns. There is no alteration in venous return to the right atrium in TOF. As blood enters the right ventricle, there is impetus for shunting across the pathway of least resistance, which in the presence of RVOT obstruction will be right to left across the VSD. Hence, in TOF, there is a greater quantity of blood—a greater stroke volume—ejected into the aorta than normal and less blood ejected into the main pulmonary artery with every beat. This may partly explain the larger than normal size of the aorta in TOF.
In the normal fetal circulation, only 15% to 20% of the right ventricular cardiac output flows into the pulmonary circulation. The majority of blood ejected from the right ventricle bypasses the lungs by crossing the DA and entering the descending aorta. Thus, the presence of RVOT obstruction in utero as seen in TOF can substantially affect the amount of blood that crosses the DA. In the presence of mild RVOT obstruction, a lesser volume of blood flow will cross the DA antegrade (from pulmonary artery to aorta). In general, in TOF, the DA is smaller than normal. In severe pulmonary stenosis or pulmonary atresia, retrograde flow in the DA (aorta to pulmonary artery) can be readily visualized prenatally. The DA often originates from a more vertical orientation adjacent to the underside of the aortic arch or takes a tortuous course, most likely as a consequence of the altered blood flow patterns . Rarely, the DA may be absent altogether.
The degree of RVOT obstruction will influence to what extent the fraction of blood reaching the pulmonary circulation is reduced. Pulmonary perfusion in severe cases of narrowing may receive blood from two sources—antegrade from the right ventricle and retrograde from the DA. In TOF, pulmonary vascular development can be abnormal. Proximal branch pulmonary arteries are smaller than normal and stenosis or narrowing is common. In some cases, small pulmonary vessels may be abnormal and, overall, the pulmonary vascular cross-sectional area can be reduced. Whether this is acquired in utero, secondary to alterations in fetal blood flow patterns and diversion of blood away from the developing lungs, or a primary finding present early at formation simply as an anatomical continuation of RVOT obstruction is unclear.
Cerebral blood flow patterns have been demonstrated to be abnormal in fetuses with TOF, which may in part be related to the alterations in blood flow patterns in the aorta. Newborns with TOF also have smaller anthropometric measurements, including head circumference. In the absence of extracardiac or genetic/chromosomal abnormality, placental function in TOF is believed to be normal.
Prenatal Management
If extracardiac abnormalities are not present, the fetus with TOF is typically stable from a hemodynamic standpoint. Postnatally, the degree of hypoxemia correlates to the degree of severity of RVOT obstruction. Because the pulmonary blood flow demands are limited prenatally, it can be a challenge to accurately predict the magnitude of postnatal hypoxemia based on fetal echocardiography. Moreover, even a mild degree of pulmonary stenosis identified at 20 weeks’ gestation can progress to more severe stenosis or atresia by late gestation, which underscores the importance of serial fetal echocardiograms. In general, a fetus with a pulmonary valve annulus that is smaller than half the size of the aortic valve annulus will likely have significant hypoxemia at birth. In addition, retrograde flow in the DA is a marker of insufficient flow across the RVOT.
Given the high frequency of associated extracardiac abnormalities, a comprehensive obstetrical ultrasound scan should be performed. The presence of a thymus or a right aortic arch increases the risk of having 22q del. Increased nuchal translucency has been found in nearly half of fetuses with TOF. An amniocentesis should be recommended to every mother carrying a fetus with TOF, because the overall prognosis and tone of counseling to families is strongly linked to the coexistence of extracardiac and chromosomal abnormalities.
Postnatal Physiology
In TOF, there is impediment to forward blood flow from the right ventricle into the pulmonary circulation. Once a baby is detached from fetal circulation, pulmonary vascular resistance decreases and pulmonary blood flow increases dramatically. However, if there is proximal blockage of blood flow as in TOF, blood will take the pathway of least resistance, which may be across the VSD to the left ventricle. Whereas, in utero, such shunting has no impact upon oxygenation, now that the fetus is on its own and separated from placenta, any blood diverted away from the lungs is deoxygenated blood and will be ejected into the aorta, leading to cyanosis.
In patients with a modest degree of obstruction, only a mild degree of hypoxemia may be present with arterial oxygen saturations measured by pulse oximetry of greater than 90%. Alternatively, patients with more significant obstruction will have a deficiency of pulmonary blood flow and develop more significant hypoxemia. The presence of a DA allows for left-to-right shunting (aorta to pulmonary artery), which will augment pulmonary blood flow. Hence, some newborns with even severe RVOT obstruction, but a patent DA, may have adequate oxygenation and saturation levels greater than 90%.
The physical examination of a newborn with TOF reveals a harsh crescendo-decrescendo systolic murmur along the left sternal border, and the intensity of the murmur correlates inversely with the degree of RVOT obstruction (the harsher the murmur, the more blood going across the obstructed region; the softer the murmur, the less blood flow across the obstruction). Varying degrees of hypoxemia will be present depending upon the amount of pulmonary blood flow. Digital clubbing is not appreciated in the newborn period but may be present in older, unrepaired infants and children.
Infants with TOF may also be at risk for intermittent episodes of severe hypoxemia, known as “hypercyanotic spells” or “tet spells.” The abrupt change in physiology is related to an increase in right-to-left shunting through the VSD, believed to be related to an acute change in the ratio of pulmonary to systemic vascular resistance. Although short-term therapies to increase systemic vascular resistance may temporarily increase pulmonary blood flow, the occurrence of hypercyanotic spells is an indication to proceed with surgical repair.
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


