CHAPTER 47 Tetralogy of Fallot
PREVALENCE AND EPIDEMIOLOGY
Tetralogy of Fallot is the most common congenital heart disease manifesting with cyanosis. The incidence is 0.06% of live births, and it constitutes 5% to 7% of all congenital heart disease.1,2 Genetic syndromes are associated with tetralogy of Fallot in 20% of cases with 22q11 deletion and trisomy 21 being the most common.3 Complete atrioventricular septal defects are present in 2% of patients with tetralogy of Fallot, especially patients with trisomy 21.4 Alagille syndrome, VACTERL syndrome (vertebral anomalies, anal atresia, cardiovascular anomalies, tracheoesophageal fistula, esophageal atresia, renal anomalies, limb anomalies), and CHARGE syndrome (coloboma of the eye, heart defects, atresia of the choanae, retardation of growth and development, genitourinary abnormalities; ear abnormalities and deafness) are also associated with tetralogy of Fallot. Atrial septal defect is present in one third of patients with tetralogy of Fallot. Currently, 85% of children with tetralogy of Fallot survive to adulthood, increasing the prevalence of tetralogy of Fallot in adults.
Aberrant subclavian arteries are associated in 5% to 8% of patients with tetralogy of Fallot. The prevalence of tetralogy of Fallot and anomalous subclavian artery is greater if associated with a right aortic arch or pulmonary atresia. Anomalous subclavian artery occurs in 24% of patients with tetralogy of Fallot, right aortic arch, and pulmonary atresia.5
Coronary artery anomalies are common, occurring in 5% to 12% of children with tetralogy of Fallot.6,7 Coronary artery anomalies that result in major branches coursing anterior to the right ventricular outflow tract (RVOT) interfere with transannular patch angioplasty of the outflow tract.
MANIFESTATIONS OF DISEASE
Clinical Presentation
A child with a palliative shunt has a continuous murmur similar to that heard in a child with a patent ductus arteriosus. After corrective surgery, symptoms may reflect continued right-sided obstruction or pulmonary regurgitation. Corrective surgery for tetralogy of Fallot requires enlargement of the RVOT and frequently a transannular patch, which results in pulmonary regurgitation.8–10 Pulmonary regurgitation is initially well tolerated, but later is associated with fatigue. The right ventricle and its outflow tract progressively dilate. Right ventricular enlargement leads to distortion of the tricuspid valve annulus and tricuspid regurgitation, compromising right heart function further. ECG abnormalities with lengthening of the QRS complex can lead to arrhythmia that may result in sudden death. Diastolic dysfunction can progress to irreversible systolic dysfunction.8 Revision of the RVOT with a valved conduit causes right ventricular remodeling and improved cardiac function. Pseudoaneurysms of the RVOT can follow corrective surgery with a transannular patch or pulmonary conduit.
Aortic root dilation and regurgitation are common in adults with tetralogy of Fallot and can lead to arterial medial abnormalities. Risk factors for aortopathy include patients with pulmonary atresia, right aortic arch, history of aorticopulmonary shunts, male sex, and chromosome 22q11 deletions.11
Imaging Technique and Findings
Radiography
Radiography plays a secondary role to echocardiography. The cardiac silhouette is frequently normal in size and configuration (Fig. 47-1). The classic radiographic appearance is a boot-shaped heart caused by elevation of the cardiac apex from right ventricular hypertrophy with a diminished or unapparent pulmonary artery segment (Fig. 47-2). The appearance of a boot-shaped heart can be mimicked if the central x-ray beam is caudal to the heart causing cephalic projection of the more anterior portion of the heart (Fig. 47-3). The pulmonary vascularity is diminished, but may be undetectable on a conventional radiograph if right ventricular obstruction is mild. A right aortic arch is visible in 25% of patients with tetralogy of Fallot. Direct visualization of the aortic arch and tracheal shift can be difficult to detect in infants and young children. The side of the aortic arch is reliably determined, however, by noting the increased density of the pedicles on the side of the aortic arch. Patients with tetralogy of Fallot and absent pulmonary artery have enlargement of the central pulmonary arteries (Fig. 47-4).
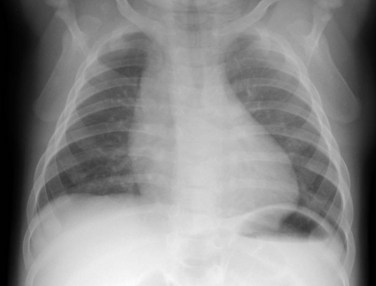
 FIGURE 47-1
FIGURE 47-1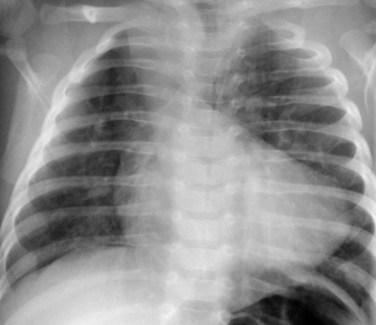
 FIGURE 47-2
FIGURE 47-2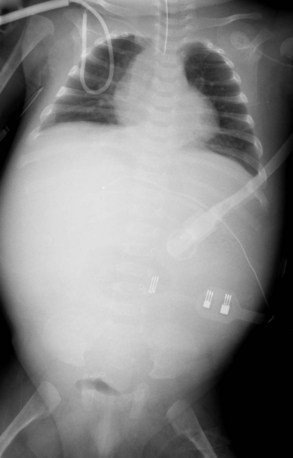
 FIGURE 47-3
FIGURE 47-3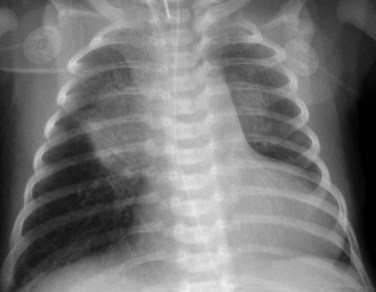
 FIGURE 47-4
FIGURE 47-4Ultrasonography
Echocardiography is definitive for the prenatal and postnatal diagnosis of tetralogy of Fallot.12 A large VSD is identified in utero on the four-chamber view, but evaluation of the outflow tracts is necessary for the diagnosis because other conotruncal abnormalities can have a VSD and an overriding aorta. A large aorta overriding the VSD is present. The aorta is larger than the pulmonary artery. Doppler interrogation of the fetal RVOT shows elevated velocity. A left-to-right shunt through the ductus arteriosus suggests severe RVOT obstruction.13
Postnatal echocardiography characterizes all intracardiac features of tetralogy of Fallot. The VSD and aorta override are identified on the parasternal long-axis view (Fig. 47-5). The outflow tract must also be evaluated (Fig. 47-6) because truncus arteriosus and VSD with pulmonary atresia can have the same appearance. In contrast to double outlet right ventricle, the aortic override to the right ventricle is less than 50%, and aortic valve fibrous continuity with the mitral valve is preserved. The RVOT including the infundibulum, pulmonary valve, and pulmonary artery is measured for stenosis. The measured infundibular volume is small, and owing to reduced growth compared with somatic growth becomes progressively more stenotic.14 Infants who require earlier surgical intervention have a greater reduction in infundibular volume. The pulmonary annulus can be hypoplastic and bicuspid. Hypoplasia with a z score less than 2 is an indication for a transannular patch. Branch pulmonary artery continuity and size is also determined. The position of the coronary arteries must be established. A coronary anomaly with a major coronary branch coursing anterior to the RVOT must be excluded before repair. CT or MRI may be necessary if the coronary arteries or pulmonary arteries are inadequately visualized.
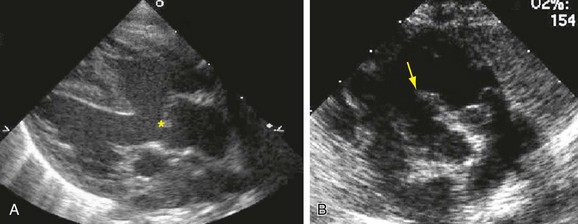
 FIGURE 47-5
FIGURE 47-5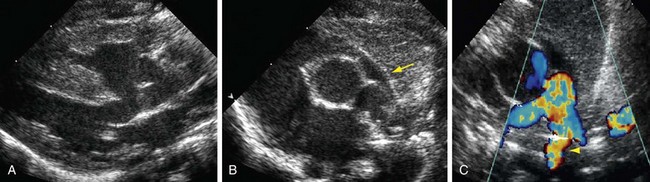
 FIGURE 47-6
FIGURE 47-6


