The Congenital Malformation Syndromes: Osteochondrodysplasias, Dysostoses, and Chromosomal Disorders
Lee F. Rogers
Sam T. Auringer
L. F. Rogers: Department of Radiology, Wake Forest University School of Medicine, Winston-Salem, North Carolina 27157. S. T. Auringer: Departments of Radiology and Pediatrics, Wake Forest University School of Medicine, Winston-Salem, North Carolina 27157.
Congenital abnormalities of bone are relatively common. It has been estimated that approximately 3% of newborns have malformations and 1% have multiple malformations.26 Some abnormalities are so common they may be considered as variations of normal. These and minor congenital anomalies are covered in Chapter 8. In this chapter, the congenital malformation syndromes are described. The term malformation syndrome refers to a constellation of abnormalities that frequently occur together.26 The syndromes are often referred to by an eponym, named for those who originally described the abnormality (e.g., Hurler’s syndrome). Alternatively, they are designated by a series of Latin or Greek phrases that include the principal sites of abnormalities. An example is the acrocephalosyndactyly syndrome (Apert’s syndrome), which consists of craniofacial, hand, and foot abnormalities. As is often the case, this syndrome is known by both types of names.
The precise cause of most malformation syndromes is unknown.20,24,26 A few, such as the mucopolysaccharidoses (i.e., Hurler’s syndrome), are known to result from specific abnormalities in metabolism, and a small number have been related to chromosomal abnormalities such as trisomy 21 (Down’s syndrome). Advances in genetic science have disclosed a number of chromosomal abnormalities, and more are anticipated.
Osteochondrodysplasias are characterized by generalized abnormalities of cartilage or bone growth.25 Dysostoses are malformations of individual bones that may occur singly or in combination; they differ from the osteochondrodysplasias in that they are focal and not generalized.
Although the malformation syndromes present a complex, complicated, rather bewildering array of seemingly endless variation, it is possible to develop a systematic approach that will, under most circumstances, lead to the correct diagnosis. There is a common misconception that most of these syndromes can be diagnosed by chemical analysis of enzymes or by chromosomal studies. In fact, most diagnoses are based on the morphology—that is, the clinical appearance of the individual patient—or on the radiographic appearance of the skeletal system. In many cases, the radiographic examination of the skeleton is essential for accurate diagnosis.
A precise diagnosis is necessary to determine the prognosis, to offer genetic counseling, and to alert the physician to the presence of associated visceral abnormalities. Certain abnormalities are lethal either in the neonatal period or shortly thereafter. Many are associated with dwarfism or mental retardation. Several skeletal malformation syndromes are associated with congenital abnormalities of the heart, usually septal defects, and some with lenticular or retinal abnormalities affecting sight. Genetic counseling is necessary for those parents who have given birth to a child with a malformation syndrome or who have a family history of malformation syndromes.
In general, evaluation of congenital malformation syndromes requires a thorough assessment of the clinical history and clinical findings, as well as a total skeletal survey.20 The diagnosis of many malformation syndromes can be established in utero by sonography.11,13 The radiographic examination of patients with a suspected malformation syndrome should include anteroposterior (AP) and lateral films of the skull and entire spine; AP films of the trunk, including the pelvis and both extremities; and a separate AP examination
of the hands and feet. This allows evaluation of the length of bones and other important characteristics. It is important to realize that not all of the findings are present in each case. By the same token, if one is attempting to confirm a clinical diagnosis and significant radiographic findings are identified that are not included in the description of the disorder being considered, it is very likely that the clinical diagnosis is incorrect and that one is dealing with a different syndrome.
of the hands and feet. This allows evaluation of the length of bones and other important characteristics. It is important to realize that not all of the findings are present in each case. By the same token, if one is attempting to confirm a clinical diagnosis and significant radiographic findings are identified that are not included in the description of the disorder being considered, it is very likely that the clinical diagnosis is incorrect and that one is dealing with a different syndrome.
RADIOGRAPHIC FEATURES OF MALFORMATION SYNDROMES
The following is a list of important considerations in the radiographic evaluation of malformation syndromes:
Relative lengths of various bones. Are the bones too short or too long? Is there some form of dwarfism? If so, is the trunk normal with affected extremities, or vice versa? If the extremities are affected, are the distal bones affected more than the proximal bones, or vice versa? Skeletal dysplasias may be identified in utero by ultrasonographic detection of short limbs and other characteristic features, both morphologic and skeletal (Fig. 9-1).19,22
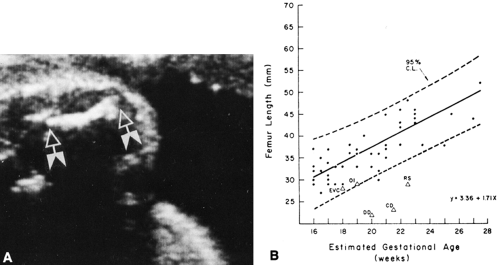
FIG. 9-1. A: Intrauterine ultrasonic detection of short limbs. The femur (arrows) measured two standard deviations below the expected length for the age. The fetus proved to have thanatophoric dysplasia. B: Femur lengths of normal fetuses in utero measured by ultrasonography. Values from fetuses with skeletal dysplasia plotted on normal curve. EVC, chondroectodermal dysplasia or Ellis-van Creveld syndrome; OI, osteogenesis imperfecta; DD, diastrophic dysplasia; CD, camptomelic dysplasia; RS, Robert’s syndrome. (From
Hobbins JC, Bracken MB, Mahoney MJ: Diagnosis of fetal skeletal dysplasias with ultrasound. Am J Obstet Gynecol 142:306, 1982, with permission.)
Involvement of the spine. There is a major group of anomalies associated with abnormalities of the vertebrae.
Age at onset. Abnormalities seen in early infancy can be separated from those seen in later life.
Fusion of bones. Fusion of various bones may be diagnostic in certain conditions. Fusion of carpal or tarsal bones can occur as a sporadic anomaly or in association with various syndromes. Normal persons may have fusions in the hand, usually involving carpals in the same proximal or distal row (see Fig. 8-3 in Chapter 8), whereas fusion of carpal bones between rows is usually associated with a malformation syndrome. However, fusions in congenital malformation syndromes may also involve carpals in the same row. In the foot, the syndrome-associated fusions usually involve the distal portions of the forefoot, which may be fused to each other or to the metatarsals. This type of fusion almost never occurs as an isolated anomaly. Fusion of proximal tarsals, such as the calcaneus and navicular (see Fig. 8-40 in Chapter 8), is often isolated and not syndrome associated, but it may be seen in syndromes as well.
Appearance of the epiphyseal complex: epiphysis, physis, and metaphysis. Alterations in the epiphyseal complex are extremely important in the characterization of various abnormalities. In adult life, many of these findings disappear and the diagnosis is more difficult. In the neonate, diagnosis of disorders affecting the epiphyses may be difficult because very few are ossified.
Appearance of the diaphysis. Is the diaphysis thick or thin? Is the cortex abnormal? Is there bowing?
Abnormalities in the number of digits. Polydactyly or decreased number of digits is seen in a variety of conditions and may be a clue to the diagnosis.
Symmetry of abnormalities. Generally, symmetrical anomalies (e.g., polydactyly, absence of digits) are associated with a familial inheritance, whereas unilateral changes are more likely to be sporadic and not part of a major syndrome.
Abnormalities of density. Are the bones too dense or too lucent?
Skeletal maturation. In the majority of syndromes skeletal maturation is delayed so that it is, in itself, of little diagnostic value. However, advanced skeletal maturation can be an important clue to diagnosis.
Pattern of anomalies. Many of the individual malformations are nonspecific. However, when associated with other anomalies, they suggest a diagnosis. Triangulation is important in establishing the diagnosis. For instance, a child is noted to have multiple carpal bones and a dislocated hip. By examining a listing of syndromes associated with multiple carpal bones and a separate listing of multiple dislocations, it is noted that each of these conditions is present in several disorders but the combination of both is found only in Larsen’s syndrome. Such listings are available in the texts by Poznanski15 and Taybi and Lachman.26 Several texts include full descriptions of the malformation syndromes and serve as excellent references; these include the works of Spranger and colleagues24 on the bone dysplasias, Taybi and Lachman26 on radiology syndromes and metabolic disorders, and Poznanski15 on the hand in radiologic diagnosis.
Change with time. The radiographic appearance of many dysplasia syndromes varies with age. The age dependence of the radiologic phenotype is a potential source of considerable diagnostic difficulty: textbook-like features in one phase of life may be completely absent in another. The atlas of Spranger and associates24 is a great aid in this regard because it demonstrates the chronologic changes in many conditions.
NOMENCLATURE OF THE CONGENITAL MALFORMATION SYNDROMES
The designations of the congenital malformation syndromes used here are those adopted by the Committee for the International Nomenclature of Constitutional Diseases of Bone (1992).23 The nomenclature was adopted to minimize problems resulting from the description of many syndromes under various names, and it is periodically revised to unify the terminology used in different parts of the world. It is not intended to be a classification of skeletal disorders.
OSTEOCHONDRODYSPLASIAS
Osteochondrodysplasias are generalized abnormalities of growth and development of cartilage or bone.8 They are subdivided into those identifiable at birth and those identifiable in later life. The other two principal categories are (1) disorders with disorganized development of cartilage and fibrous components of the skeleton and (2) abnormalities of density of cortical or diaphyseal bone structure.
Defects in Growth of Tubular Bones or Spine Identifiable at Birth
Achondroplasia
Achondroplasia is the most common form of dwarfism. It has been recognized since antiquity and has been commonly identified with court jesters and with clowns in circuses. It is an hereditary congenital disturbance transmitted as an autosomal dominant trait that causes inadequate endochondral bone formation and results in dwarfism. Membranous bone formation is not affected. The mental status is normal.
Characteristically, the short limb bones contrast with the normal length of the trunk. The facies is characterized by prominent frontal bossing of the skull, saddle nose, and prognathic jaw. All achondroplastic persons bear a strong anatomic resemblance to each other. The following paragraphs describe the most typical changes in the skeleton.18,24
Roentgenographic Findings
Long Tubular Bones
Shortening of the long bones is responsible for dwarfism (Figs. 9-2 and 9-3). The humerus
and femur tend to be relatively more affected than the distal bones of the extremities (rhizomelic shortening). The diameter is usually normal, but the bones appear thick because they are short. The ends of the shafts are flared. The zone of provisional calcification may be smooth or irregular. At times there is a sizable V-shaped notch in the metaphyses, and the epiphyseal centers may be partially buried in the metaphyses; this is known as the ball-in-socket epiphyseal-metaphyseal junction (see Fig. 9-2). The fibula often is longer than the tibia, causing an inversion of the foot (Fig. 9-3). Bowing of the long bones is common. The diagnosis can be established in utero by sonography.11,13
and femur tend to be relatively more affected than the distal bones of the extremities (rhizomelic shortening). The diameter is usually normal, but the bones appear thick because they are short. The ends of the shafts are flared. The zone of provisional calcification may be smooth or irregular. At times there is a sizable V-shaped notch in the metaphyses, and the epiphyseal centers may be partially buried in the metaphyses; this is known as the ball-in-socket epiphyseal-metaphyseal junction (see Fig. 9-2). The fibula often is longer than the tibia, causing an inversion of the foot (Fig. 9-3). Bowing of the long bones is common. The diagnosis can be established in utero by sonography.11,13
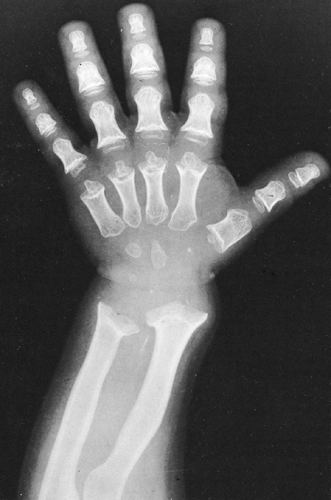 FIG. 9-2. Achondroplasia in a child. The bones of the hand are short and broad; there are ball-in-socket epiphyseal-metaphyseal junctions at the distal ends of the metacarpals. Note the flaring of the metaphyses, particularly of the radius and ulna. |
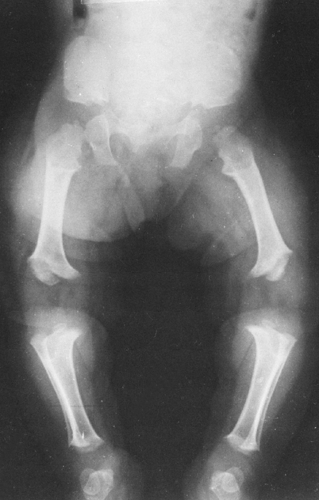 FIG. 9-3. Achondroplasia in an infant. The shortening of the femur is more pronounced than that of the tibia. Note that the length of the fibula is proportionally greater than that of the tibia. The distal femoral metaphyses are tilted upward and laterally, with laterally placed epiphyses. Typical changes are present in the pelvis. |
Short Tubular Bones
These bones show changes similar to those seen in the long bones. They are short and appear thick, and the fingers tend to be of similar lengths, the so-called trident hand (see Fig. 9-2).
Pelvis
Characteristic changes occur in the pelvis. The ilia are short and square, and the sacrosciatic notch is small. The ischial and pubic bones are also short and broad. The acetabular angles are decreased in infancy. The sacrum articulates low on the ilia (Fig. 9-4).
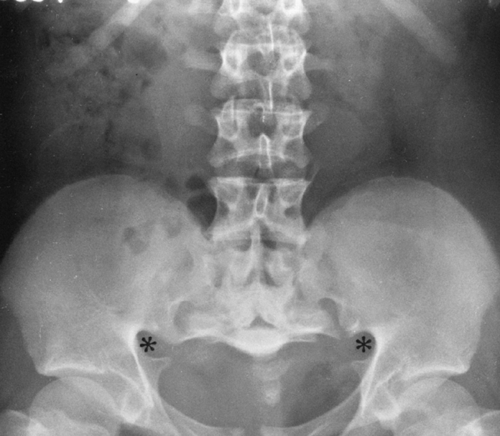 FIG. 9-4. Achondroplasia in an adolescent. The ilia are short and square, and the acetabular angles are flat. The pelvis is foreshortened in the anteroposterior direction, and there are characteristic deformities of the sacrosciatic notches (asterisks). The sacrum is horizontal and projected on end. The interpediculate distance narrows progressively from the upper to the lower lumbar spine, the reverse of normal. |
Spine
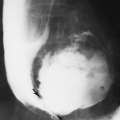
The length of the spinal column may be normal or nearly so, but vertebral development is affected. One or several vertebrae at the thoracolumbar junction (T12 to L3) may be rounded or wedged anteriorly. In the lumbar area, the interpedicular distances characteristically narrow progressively from above downward (see Fig. 9-4), the reverse of the normal. The pedicles are short and thick, and the posterior surfaces of the bodies are concave (Fig. 9-5). The net effect is a stenotic spinal canal, which in the adult may lead to severe neurologic symptoms, especially if a herniated disc or degenerative joint disease or kyphosis develops (Fig. 9-6). The lumbar lordosis is increased, and the lumbosacral angle becomes more acute than is normal (Fig. 9-5). The long axis of the sacrum tends to be horizontal.
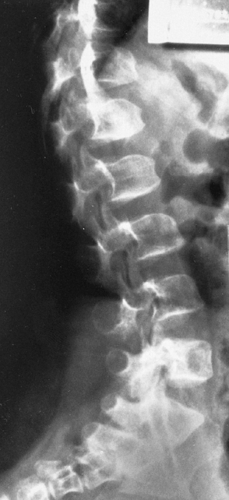 FIG. 9-5. Achondroplasia in a 6-year-old. The vertebrae at the thoracolumbar junction are rounded and are wedged anteriorly and concave posteriorly. The pedicles are short. The lumbosacral angle is accentuated by the horizontal orientation of the long axis of the sacrum. |
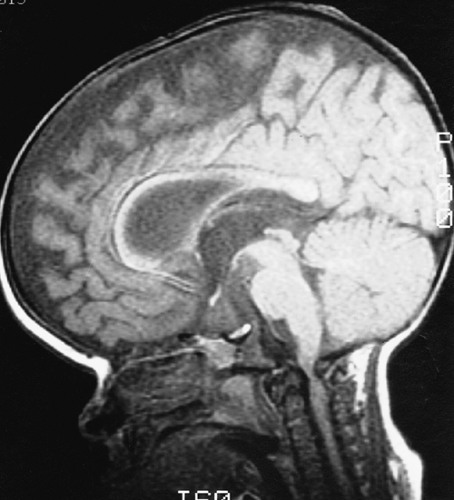 FIG. 9-6. Achondroplasia. Sagittal magnetic resonance image demonstrates a severely stenotic foramen magnum with conus compression. |
Skull
The skull is brachycephalic because the base of the skull is preformed in cartilage and therefore hypoplastic. The vault is relatively large, with bulging of the frontal bones. It is of membranous origin and therefore is not affected. The growth of the mandible is relatively normal and therefore appears prognathic.
Other Bones
The carpal and tarsal bones are normal. The scapula is short, and the sternum may be thick and short. The ribs are short, causing a decrease in the AP diameter of the thorax.
Thanatophoric Dysplasia
Thanatophoric dysplasia was previously mistaken for a severe type of achondroplasia because of its many similar features. These similarities include dwarfing of the long tubular bones with a relatively long trunk; a prominent forehead with a short skull base and depression of the nasal root; small, square iliac wings with horizontal acetabular roofs; a narrow thorax with short ribs; and a decrease from above downward of the interpedicular distances in the lumbar spine. Thanatophoric dysplasia is the most common lethal neonatal skeletal dysplasia. Distinguishing features include Kleeblattschädel or cloverleaf skull; severe platyspondyly or extremely thin, wafer-like ossification of vertebral bodies with thick intervertebral spaces; very short limb bones with bowing, especially in the lower extremities (also known as French telephone receiver femurs); and no history of a similar disorder in other members of the family (Fig. 9-7).
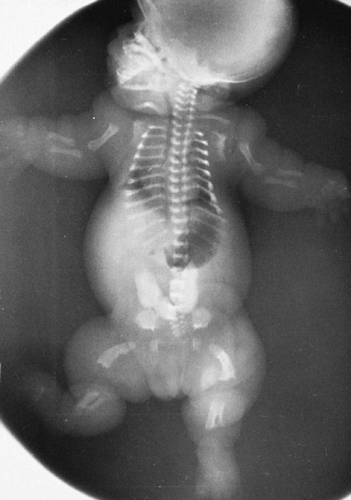 FIG. 9-7. Thanatophoric dysplasia. The limb shortening is more pronounced proximally than distally. Note the narrow thorax with short ribs and characteristic flattening of the vertebral bodies. The pelvis is deformed in the characteristic pattern. The femurs are short and bowed. |
Affected infants are stillborn or die shortly after birth, possibly from respiratory failure owing to the short ribs and narrow thorax.
Short ribs with a narrow thorax occurs in three principal dysplasias—thanatophoric dysplasia, asphyxiating thoracic dystrophy, and achondroplasia. Other changes common to achondroplasia may also be seen in the other two abnormalities. A small chest is also a feature of the short rib polydactyly syndromes.26
Asphyxiating Thoracic Dystrophy
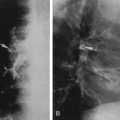
Asphyxiating thoracic dystrophy was first reported by Jeune in 1955 in two infant siblings who died of respiratory distress associated with small and relatively immobile thoraces. The major abnormalities identifiable on roentgenography consist of very short ribs and a variable degree of shortening of the long tubular bones with metaphyseal notching.
The ribs project horizontally and may be so short as to barely reach the anterior axillary line (Fig. 9-8). The shortened ribs reduce the volume of the thorax and are responsible for the respiratory distress. The cardiac silhouette often appears large, but this is probably an illusion because of the smallness of the thorax. In the pelvis, the ilium is shortened in its inferosuperior diameter, the acetabular roof is broad, and there may be a deep V-shaped notch in it. The disease may be fatal, with the infant dying from respiratory complications. Associated renal disease has also been reported, and renal failure may be the cause of death in patients with less severe skeletal changes.
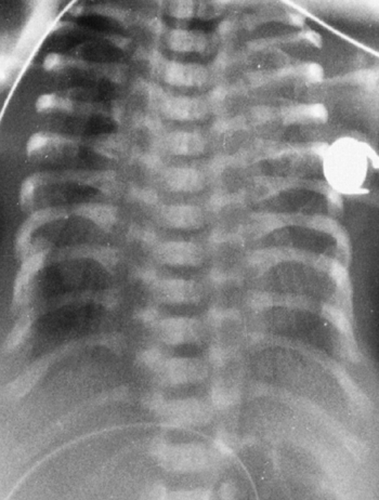 FIG. 9-8. Asphyxiating thoracic dystrophy. The thorax is extremely small and the ribs quite short. The cardiac silhouette appears large because of the smallness of the thorax. The vertebral bodies are larger and the intervertebral disc space is narrower than is the case in thanatophoric dysplasia. |
Diastrophic Dysplasia
Diastrophic dysplasia is inherited as an autosomal recessive trait. The syndrome is characterized by the combination of scoliosis and clubfeet. There is also a delay in appearance of the epiphyseal centers and subluxation of various joints (especially the hips).
The long bones are short and thick, with widened metaphyses, simulating achondroplasia. In the hands, the bones are short, especially the thumb, which may project at a right angle to the other digits, the so-called hitchhiker’s thumb (Fig. 9-9). The metacarpal of the thumb also may have an ovoid shape. The bones of the feet show changes similar to those in the hands. In addition, there is a bilateral clubfoot deformity.
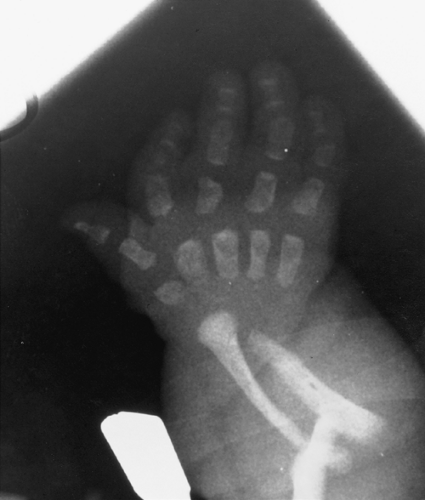 FIG. 9-9. Diastrophic dysplasia. The “hitchhiker’s thumb” is demonstrated with its short and ovoid first metacarpal. Note the short proximal phalanges and metacarpals. |
The epiphyseal centers are late in appearing and, when they do appear, are apt to be flat and abnormal in shape. In the hand, the epiphyseal centers may be oriented parallel rather than perpendicular to the long axis of the phalanges. That is, the height is longer than the width, the reverse of the normal proportion. In most patients, laxity of ligaments and tendons causes subluxation of the joints. The subluxations are not present at birth but develop after the child begins to walk. Scoliosis and kyphosis also appear at about the same time. A severe kyphosis in the cervical region can be fatal in infancy. Other bones, including the vertebrae, skull, and pelvis, are normal. The tarsal bones may be distorted because of the equinovarus deformity but otherwise are normal.
Chondrodysplasia Punctata (Stippled Epiphyses)
There are 15 described types of chondrodysplasia punctata,14 and the disease is genetically transmitted with both dominant and recessive or rhizomelic forms. Infants with the rhizomelic or recessive forms are frequently stillborn or die within the first year of life from associated abnormalities or intercurrent disease.
The characteristic roentgenographic finding is the presence of numerous small, round opacities in the unossified epiphyseal cartilages (Fig. 9-10). In some patients, the opacities appear to extend into the adjacent soft tissues. They also have been found in other cartilages such as the nasal septum, larynx, and trachea. Stippling of the vertebral cartilages is common in the more severe cases. The extremities may be dwarfed, and flexion deformities may also be present. The femur and humerus are most likely to be shortened in the recessive types. Many patients have congenital cataracts, saddle nose, hyperkeratotic dermatoses, and failure of proper mental and physical development. If the infant survives, the foci may ossify and then merge to form a fairly normal epiphyseal center in some cases. The fingers and toes may be short and stubby. The carpal and tarsal bones may be normal or show some irregularity in shape.
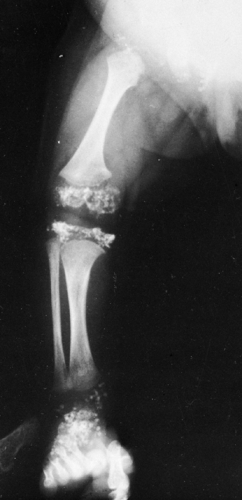 FIG. 9-10. Stippled epiphyses. Numerous tiny, dense foci were noted throughout the cartilages of the skeleton. They were most numerous in the right lower extremity. The bones are shorter than normal. (From Paul LW: Punctate epiphyseal dysplasia (Chondrodystrophica calcificans congenita) Am J Roentgenol 71:941, 1954 . Copyright 1954 American Roentgen Ray Society, with permission.) |
Stippled epiphyseal calcification has also been noted in the Zellweger or cerebrohepatorenal syndrome, other forms of chondrodysplasia punctata, warfarin embryopathy, and alcohol embryopathy.14
Chondroectodermal Dysplasia (Ellis-van Creveld syndrome)
Chondroectodermal dysplasia was first described among the Old Order Amish People of Pennsylvania. The disease
is transmitted as an autosomal recessive trait. Cardiac anomalies (atrial septal defects being the most frequent) have been found in about 60% of those affected. The ectodermal component of the syndrome is manifested by small, friable nails; defective dentition; and, in a few cases, alopecia.
is transmitted as an autosomal recessive trait. Cardiac anomalies (atrial septal defects being the most frequent) have been found in about 60% of those affected. The ectodermal component of the syndrome is manifested by small, friable nails; defective dentition; and, in a few cases, alopecia.
The changes in the skeleton are usually characteristic. Polydactyly and syndactyly are almost universal. A partially or completely formed sixth metacarpal may be fused to the fifth (Fig. 9-11). Fusion of the hamate and capitate bones in the wrist is often present. Cone epiphyses are common during childhood. Shortening of the long tubular bones characteristically becomes more severe distalward, the opposite of the situation encountered in achondroplasia. The tibia and fibula therefore are much shorter than the femur (mesomelic shortening), and the distal phalanges are more dwarfed than the proximal. The distal end of the radius and the proximal end of the ulna are somewhat enlarged. Also, the radial head may be flared and frequently is dislocated. The proximal end of the tibia also is widened, and the epiphysis is offset medially. Frequently, a small exostosis is present on the upper inner cortex of the tibia. The intercondylar notch of the femur is shallow, and the tibial spine is small. A severe cervical kyphosis with spinal cord compression may occur.
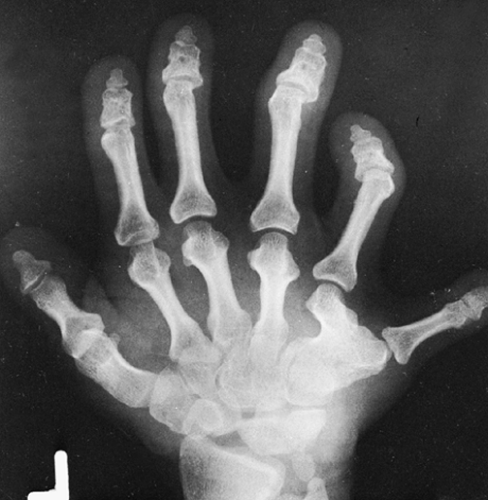 FIG. 9-11. Ellis–van Creveld disease in a woman. There is progressive shortening of the bones distally from the wrist, with polydactyly, syndactyly, and carpal fusions. (Courtesy of M. Pinson Neal, Jr., M.D., Richmond, Virginia.) |
The ribs are short. The ilia are flared and hypoplastic, with a trident deformity of the acetabular roofs. The skull and spine usually are normal.
Metatrophic Dysplasia
Metatrophic dysplasia is another form of dwarfism that may be confused with achondroplasia. Roentgenographic changes are apparent at birth, with shortening of the long tubular bones and hyperplastic, greatly flared metaphyses. There is overconstriction of the midshafts, so that the bones have a dumbbell shape (Fig. 9-12). Epiphyseal ossification
centers are delayed in appearance and, when they do appear, are deformed. The ribs tend to be short and the chest narrow. Kyphoscoliosis is present, as is platyspondyly (i.e., flattening of the vertebrae). In the pelvis, the iliac wings are short, the sacroiliac notches are short and deep, and the acetabula are horizontal. As the child grows older, there is lengthening of the long bones, more than is seen in achondroplasia. However, the platyspondyly and the kyphoscoliotic curvature worsen, and the child changes clinically from an appearance similar to achondroplasia to that resembling spondyloepiphyseal dysplasia. The base of the skull is not foreshortened, and the interpedicular distances in the lumbar spine usually remain normal, findings that aid in differentiating this dysplasia from achondroplasia during infancy.
centers are delayed in appearance and, when they do appear, are deformed. The ribs tend to be short and the chest narrow. Kyphoscoliosis is present, as is platyspondyly (i.e., flattening of the vertebrae). In the pelvis, the iliac wings are short, the sacroiliac notches are short and deep, and the acetabula are horizontal. As the child grows older, there is lengthening of the long bones, more than is seen in achondroplasia. However, the platyspondyly and the kyphoscoliotic curvature worsen, and the child changes clinically from an appearance similar to achondroplasia to that resembling spondyloepiphyseal dysplasia. The base of the skull is not foreshortened, and the interpedicular distances in the lumbar spine usually remain normal, findings that aid in differentiating this dysplasia from achondroplasia during infancy.
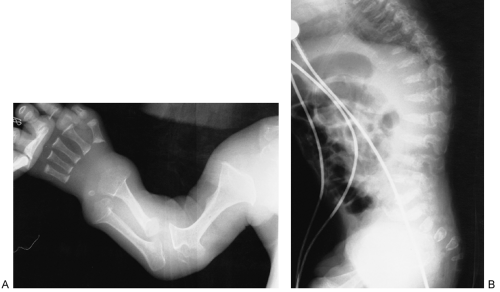 FIG. 9-12. Metatrophic dysplasia. A: The long tubular bones are short, with greatly flared, “dumbbell” metaphyses. B: The spine is kyphotic with platyspondyly. |
Spondylocostal Dysostosis
Spondylocostal dysostosis17 is a form of truncal dwarfism caused by segmental abnormalities of the entire spine (Fig. 9-13). The extremities are normal. When severe, it may lead to death in the newborn. The spinal canal is normal, but the segmentation of the vertebral bodies is entirely abnormal. The ribs are thin and often fused medially, giving a fan-like appearance. The syndrome occurs in both autosomal dominant and autosomal recessive forms and is common among Puerto Ricans.
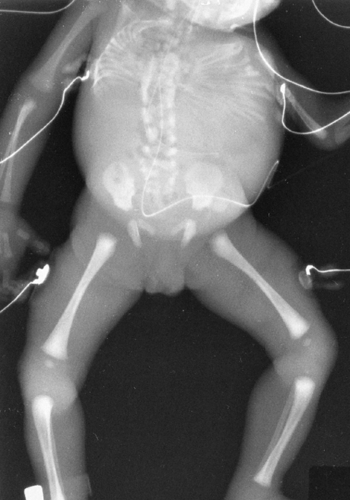 FIG. 9-13. Spondylocostal dysostosis. The trunk is shortened because of segmentation defects throughout the entire spine. The limbs are normal. Note the thin ribs with multiple interosseous fusions. |
Defects in Growth of Tubular Bones or Spine Identifiable Later in Life
Multiple Epiphyseal Dysplasia
The characteristic finding in multiple epiphyseal dysplasia is the presence of multiple ossification centers for the affected epiphyses, giving them a fragmented appearance. It is considered to be transmitted as an autosomal dominant trait with complete penetrance but variable expression. The epiphyses are usually flat, and the ends of the long bones may be somewhat flared. The involvement may be limited to a pair of epiphyses, or all epiphyses throughout the body may be involved. Symmetrical involvement of the capital femoral epiphyses is common. Knock-knee or bowleg deformity may be present, and flexion deformities at the knee joint may be seen. A coronal cleft in the patellas (double-layered patella) has been described as a fairly consistent and characteristic finding in the tarda form.
The vertebrae may be flattened in the thoracic spine, with irregular end-plates. The skull is normal. The long bones appear to be short and thick; the thickness is an illusion caused by the shortening. The carpal and tarsal bones are often irregular and the digits stubby. In many patients, however, the hands are normal.
The clinical presentation is usually limpness, pain, or stiffness in the lower extremities in late childhood or adolescence. Irregularity of the articular plates frequently persists (Fig. 9-14) and leads to the early development of degenerative joint disease, particularly in the hips. At this stage it usually is impossible to determine what the primary disorder was, because the other epiphyseal dysplasias may also result in early degenerative joint disease.
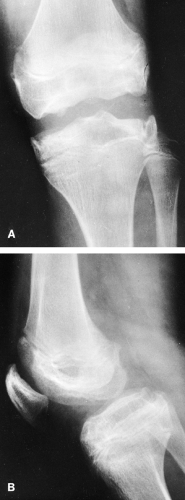 FIG. 9-14. Multiple epiphyseal dysplasia in an adolescent. The articular surfaces are seen to be irregular in both the frontal (A) and lateral (B) projections. The joint abnormality often leads to the early development of degenerative joint disease. |
When the heads of the femurs alone are involved, bilateral Perthes’ disease and the epiphyseal dysgenesis of cretinism must be considered.
Metaphyseal Chondrodysplasia, Schmid Type
In 1949, Schmid reported a mild form of metaphyseal chondrodysplasia that is relatively common. It is transmitted as an autosomal dominant trait. Irregularity of the zones of provisional calcification resembles rickets and is caused by extension of cartilage into the metaphyses (Fig. 9-15). The spine and skull are not affected. The skeletal changes are not present at birth but appear at 3 to 5 years of age. Bowing of the legs is marked, and patients have a waddling gait. There is bilateral coxa vara. The epiphyseal plates are widened. Again, the appearance may be similar to that of vitamin-D-resistant rickets. Differentiation may depend on biochemical analysis of blood and urine. Unlike rickets, the




Related posts:
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
