The External Auditory Canal and Pinna
The external ear, comprised of the pinna or auricle and the external auditory canal (EAC), has a different embryological origin than that of the middle and inner ears. This unique embryonic origin, in conjunction with the superficial location of the pinna and EAC, results in a distinct pathology spectrum when compared with that involving the middle and inner ears. Despite the ease of visualization of these external structures by the otolaryngologist, the radiologist can assist with important information for surgical planning and aid in determining the type and extent of the pathology. Intracranial complications arising from disorders of the pinna and EAC can also be evaluated by the radiologist.
Imaging Techniques
At our institution, we perform temporal bone computed tomography (CT) studies more frequently than magnetic resonance imaging (MRI) studies for examination of the EAC because of CT’s superiority for the evaluation of bone erosion. With the use of a multislice CT scanner, submillimeter (0.6 to 0.75 mm) axial images are routinely obtained and processed with bone and soft tissue algorithms. Although direct coronal imaging may be performed using a similar protocol, coronal and sagittal reconstructions can also be obtained at 0.75 mm intervals with limited radiation exposure, which is especially desirable in children. For congenital lesions, noncontrast studies with bone windows will suffice for most patients. For some inflammatory and neoplastic lesions, contrast material may be given and images processed with soft tissue window settings.
MRI is usually used to assess the spread of tumor or inflammatory processes intracranially. Although lesions that spread through bone into the middle cranial fossa can be obvious on reconstructed coronal and sagittal images, MRI is generally better for the assessment of soft tissue, brain, and dural extension. Subtle dural enhancement may be a first indication of intracranial involvement. MRI is also sensitive for detection of bone marrow edema, and it can occasionally demonstrate bony involvement not suspected by CT in carcinoma cases. Our MRI protocol includes sagittal T1-weighted images (4 mm thick), axial T1-weighted images (2 mm thick), axial T2-weighted images (2 mm thick), and axial FLAIR (fluid attenuated inversion recovery T2-weighted images, 4 mm thick), all obtained before contrast is administered. After gadolinium contrast administration, we obtain T1-weighted (3 mm thick) images in axial and coronal planes through the regions of interest with and without fat suppression techniques. Additionally, 4 to 5 mm thick axial and coronal postcontrast T1-weighted images are obtained to include the entire brain. Images using heavily T2-weighted techniques with high resolution (constructive interference in steady state; CISS) may be performed if anatomical assessment of the structures in the inner ear or internal auditory canal is required. For patients with facial paralysis, it is imperative that the imaging studies include the entire course of the facial nerves.
Anatomy
At birth, the tympanic membrane (TM), ossicles, and otic capsule are already of adult size, but the EAC grows progressively until the age of 9 years, when it achieves its typical S-shaped course and adult size. The EAC extends from the meatus of the pinna to the TM, its medial boundary, which divides the external from the middle ear. The purpose of the pinna and EAC is presumably to collect and funnel sound to the TM as a resonating tube resulting in a 10 to 20 dB gain.1 Thus, atresia and stenosis of the EAC can result in a conductive hearing loss of up to 60 dB, manifested as an air–bone gap on audiological testing. The adult EAC is ∼ 25 mm in length and air-filled in its normal state. The EAC is lined by squamous epithelium that is continuous with the skin from the pinna. The canal travels inferiorly and posteriorly to the TM in a slightly S-shaped course. The resulting cross-sectional diameter has a roughly oblique oval configuration.2 The canal is widest in diameter at its lateral/superficial end adjacent to the pinna, where it flares out in a trumpet bell-like configuration. The EAC has two portions: the lateral portion (one third) is fibrocartilaginous and demonstrates an incomplete elastic cartilage ring measuring 8 mm in length. Inferiorly in the fibrocartilaginous portion are two deficiencies called the fissures of Santorini, which increase the flexibility of the superficial portion of the EAC, but may also allow transmission of infection and malignancy.1 The fibrocartilaginous portion has thick skin with hair follicles and cerumen glands. The medial two thirds of the EAC, adjacent to the TM, is the osseous portion, which is surrounded by membranous bone and devoid of cerumen glands and hair follicles, and is lined by a thin layer of skin. The lack of thick skin and subcutaneous tissue makes this portion of the canal exquisitely sensitive to touch on clinical examination. The EAC has two areas of narrowing: one at the isthmus (between the fibrocartilaginous and bony portions) and one distally and adjacent to the TM. The medial bony portion can have an anteroinferior area of dehiscence called the foramen of Huschke. The bony portion of the EAC is encircled by the squamous and mastoid segments of the temporal bone superiorly and posteriorly while the anterior and inferior portions of the bony EAC are surrounded by the tympanic portion of the temporal bone.1
The TM is situated obliquely at the distal end of the EAC, is conical in shape, and measures 9 to 10 mm in diameter. It has a peripheral fibrocartilaginous annulus that attaches to the tympanic ring in a well-defined bony sulcus. This fibrocartilaginous ring has an area of deficiency superiorly, above the short (or lateral) process of the malleus, called the notch of Rivinus. From this notch arise anterior and posterior malleal ligaments that create folds in the TM, extending to the attachment of the lateral process of the malleus. The TM is composed of both the pars flaccida and pars tensa portions. The pars tensa is comprised of three layers: an inner mucosal epithelial layer, a central fibrous layer, and an outer squamous epithelial layer. The inner fibrous layer gives this portion of the TM a more rigid structure than the pars flaccida. The pars flaccida is substantially smaller in area than the pars tensa and is superiorly located above the short process of the malleus, covering the bony notch of Rivinius. The pars flaccida of the TM is thinner and therefore more flexible, and is the point of origin of many primary, acquired cholesteatomas (CHs) of the middle ear.3
Arterial supply to the EAC is via branches of the external carotid artery, namely, the posterior auricular, superficial temporal, and internal maxillary arteries. The venous drainage from this region is via the postauricular and superficial temporal veins into the sigmoid sinus and internal and external jugular veins.1 Lymphatic drainage is anteriorly into the preauricular nodes, posteriorly into the mastoid nodes, and inferiorly into the subparotid nodes. All of these nodal stations drain to the digastric nodes. The innervation of the external ear is from multiple somatosensory nerves, including cranial nerves (CN) V, VII, IX, and X, as well as cervical nerves C2 and C3. The trigeminal nerve (CN V) fibers arise via the mandibular division as the auriculotemporal nerve. Recent studies regarding the somatosensory contribution of CN VII to the external auditory canal have demonstrated these branches arise in the descending or mastoid portion of the facial nerve prior to CN VII’s exit from the stylomastoid foramen. The CN IX contribution, which supplies the medial aspect of the TM, arises inferiorly in the hypotympanum from the tympanic canaliculus. Similarly, CN X contributes innervation to the medial aspect of the EAC via Arnold’s nerve, which enters the temporal bone through the mastoid canaliculus and exits through the tympanomastoid suture.1
Embryology
As with all discussions of embryology, we will begin with the normal sequence of events occurring in utero. The pharyngeal (or branchial) arches arise from migration of neural crest cells by the fifth week of gestation. The first branchial or mandibular arch gives origin to the mandible, maxilla, zygomatic temporal bone, squamous temporal bone, and muscles of mastication, as well as the anterior belly of the digastric muscle, tensor tympani, tensor veli palatini, and mylohyoid muscles. Also derived from the first arch are the head and neck of the malleus and the short process and body of the incus. Derivatives from the second branchial arch include the long process of the incus and stapes superstructure, lesser horn (cornu) and upper rim of the hyoid bone, styloid process, stylohyoid ligament, muscles of facial expression, posterior belly of the digastric muscle, and stapedius and stylohyoid muscles.4 The EAC arises from the ectoderm of the first and second branchial arches and therefore is composed of tissues from both the mandibular (first) and hyoid (second) arches. The formation of the EAC occurs during the sixth week of fetal life as a cleft invaginates. By this time, the tympanic cavity has already formed from invagination of the first pharyngeal pouch (tubotympanic recess) during the fourth gestational week. The TM forms from the meeting of these two invaginations as the collection of epithelial cells between them is canalized during the 26th week of life.5
The pinna arises from the six hillocks of the first and second brachial arches along the course of the first brachial cleft. The first three hillocks form the helix and tragus, and the third through sixth hillocks form the remaining auricle. Therefore, abnormal hillock development leads to microtia or anotia of the pinna, the severity of which is related to EAC canalization.6 Abnormal accessory hillocks can be seen in some patients as preauricular tags and/or cysts (Fig. 2.1). Pinna or auricle malformations occur in 1 out of 12,500 births, and arise during the 3rd to 12th fetal week of life, with the auricle and membranous EAC developing from the same anlage. Because the bony EAC is developing synchronously but from a different anlage, maldevelopment of the bony canal can also occur.7
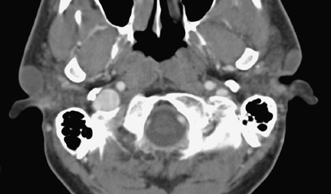
Fig. 2.1 Axial computed tomography scan shows incidentally found large bilateral preauricular cysts. These cysts may become symptomatic due to infection; however, many small ones remain clinically silent throughout the life of the patient.
Pathology
Embryologic
Microtia and External Auditory Canal Dysplasias
Microtia, meaning a small auricle, is more common in Navajo and Japanese populations as well as in fetuses exposed to thalidomide and retinoic acid.8 Microtia has three degrees of severity, according to the Weerda classification:9
- First degree: Microtia, prominent ear, pocket ear, absence of the upper helix, absence of the tragus, clefts, lobular deformities, and cup ear deformities type 1 and 2
- Second degree: Dysplasia as a cup ear deformity type 3 and mini-ear
- Third degree: Absence (i.e., anotia) of a normal auricular structure (unilateral or bilateral), or severely dysplastic ears displaced inferiorly due to incomplete ascension from the neck
In a study by Mayer et al, one third of patients had first- or second-degree microtia, and two thirds had third-degree microtia. Seventy-five percent of patients with mild microtia have associated bony or cartilaginous EAC stenosis, and 75% of those with major microtia exhibit EAC atresia.10
Maldevelopment of the first brachial arch affecting the EAC can occur syndromically or nonsyndromically and can manifest as EAC atresia or stenosis as well as duplication anomalies, including cysts, sinuses, and fistulas. These anomalies rarely occur together, but have been reported to coexist with one another occasionally.11 EAC atresia occurs in 1 out of 10,000 births and more commonly involves the lateral membranous or fibrocartilaginous portion of the EAC than its bony portion. EAC atresia may be associated with deletions in 18q, and 66% of individuals with these deletions have congenital aural atresia or EAC stenosis.12 These anomalies may also be seen in Goldenhar, Treacher Collins, Pfeiffer, and Rasmussen syndromes.13
On physical examination, the right ear is more frequently affected, and patients have a conductive hearing loss. Many of the severe cases may have a mixed hearing loss due to associated inner ear malformations. Because the EAC continues to grow during the first 2 years of life, the severity of the atresia may change during that time. Patients with EAC stenosis or atresia usually present prior to discharge from the newborn nursery because of either an obvious visible deformity of the pinna or failure on a newborn hearing screening examination (Fig. 2.2). There is a three-stage grading system for severity for EAC atresia, as follows:14
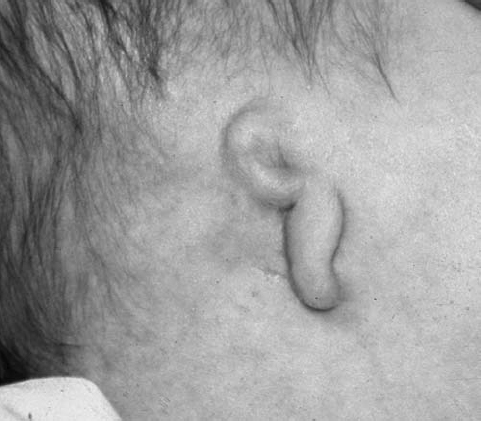
Fig. 2.2 Lateral patient photograph shows microtia with deformed residual pinna and atresia of the external auditory canal. (See Color Plate Fig. 2.2.)
- Mild EAC atresia: Normal auricle, minimal ossicular deformity, and a normal middle ear cavity
- Moderate EAC atresia: Small rudimentary pinna, small middle ear cavity, severely deformed ossicles, and an aberrant course of the seventh cranial nerve (Fig. 2.2)
- Severe EAC atresia: Absent auricle and only a small cleft for the middle ear cavity with absent ossicles. This severe form can also have inner ear deformities.
There is also a classification by location and extent of severity for EAC atresia, as follows:15
- Type A (meatal): Fibrocartilaginous only with a small TM
- Type B (partial): Fibrocartilaginous and osseous. The malleus may be fixed, or the manubrium may be short or curved
- Type C (total): The EAC is absent, and there is a bony atretic plate with a normal-size middle ear cavity. The malleus is usually fused to the lateral wall.
- Type D (hypopneumatic total): The middle ear cavity is small, and there is little or no mastoid pneumatization.
The facial nerve is aberrant (common form with mandibulofacial dysostosis).
Nonsyndromic causes of EAC stenosis and atresia are due to failure of canalization. This failure can be complete, resulting in atresia, or incomplete, resulting in stenosis (Fig. 2.3). It is well known that severe microtia is often seen with EAC atresia, whereas milder microtia is typically seen with EAC stenosis (Fig. 2.4). Associated dysplasia of the ossicles occurs in 98% of patients and in 72% of them may involve all of the ossicles, including the stapes. Accompanying round window atresia is found in 6% of patients and labyrinthine malformations in 13%, but not atresia of the oval window.10,16 Common ossicular abnormalities include a short or absent manubrium of the malleus, a more pronounced deformity of the head of the malleus than that of the incus, and misshapen stapes crura. Ossicular fusion is seen in 54% of EAC atresia patients and most commonly involves the malleoincudal joint (76%), followed by fusion of the ossicles to the atresia plate.
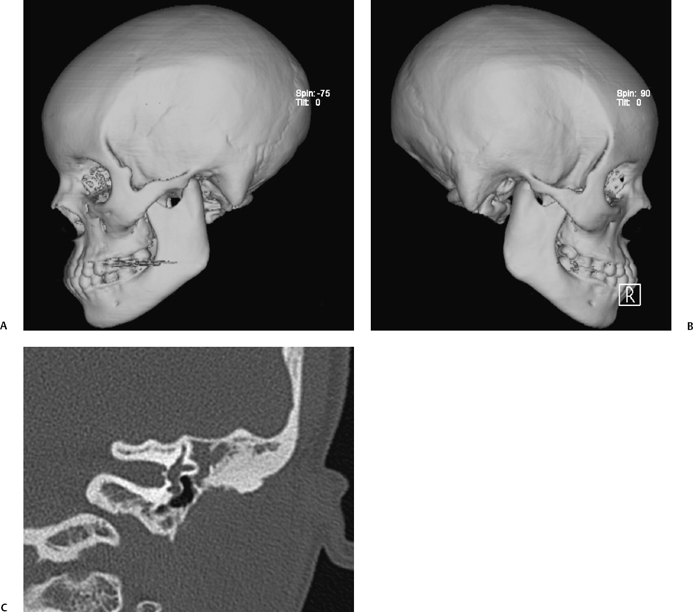
Fig. 2.3 (A,B) Computed tomography (CT) three-dimensional reformations in a patient with nonsyndromic bilateral external auditory canal (EAC) atresia. In nonsyndromic EAC atresia, the zygomatic arch and mandible are normally formed, whereas they may be hypoplastic or even absent in syndromic EAC atresia. (C) Coronal CT view shows a thin atresia plate, small middle ear cavity, absent ossicles, and soft tissue filling the epitympanic space.
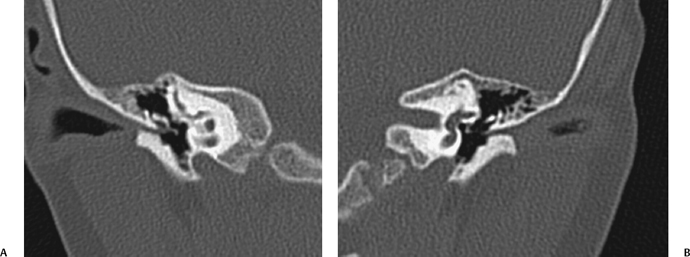
Fig. 2.4 (A) Coronal computed tomography (CT) scan shows severe right-sided soft tissue and distal bony external auditory canal stenosis. The long process of the incus is mildly deformed but not fused. The size of the middle ear cavity is normal. (B) On the left, CT shows similar findings in this patient with bilateral microtias.
The atresia plate may be bony or membranous (soft tissue) and of variable thickness (Fig. 2.5). Also, the middle ear cavity may be small (68%), and the facial nerve may be displaced.17,18 Less commonly, accessory ossicles can be seen with a type 2 first branchial cleft anomaly.19 The association of incus and malleus abnormalities with EAC stenosis/atresia occurs due to the common (i.e., branchial) embryologic origin of all of the ossicles except for the stapes footplate. EAC atresia and mallear manubrium development are related, a finding that has been substantiated in a knockout mouse model.20 Also associated with microtia and EAC atresia are poor pneumatization of the mastoid, mandibular condyle dysplasia, zygomatic arch defects (50%), eustachian tube dysplasia (20 to 40%), tensor tympani muscle hypoplasia (20 to 40%), oval window absence (33%), labyrinthine dysplasia (13%), and/or round window absence (6%). Associated hypoplasia or aplasia of the internal carotid artery is rare.10 There can be associated CH of the middle ear medial to the atresia plate. More commonly, CH can develop in the EAC medial to a stenosis (Fig. 2.6). The presence of a CH correlates with smaller EAC size, so that the rate of CH is 50% if the EAC is 4 mm or less in diameter.21
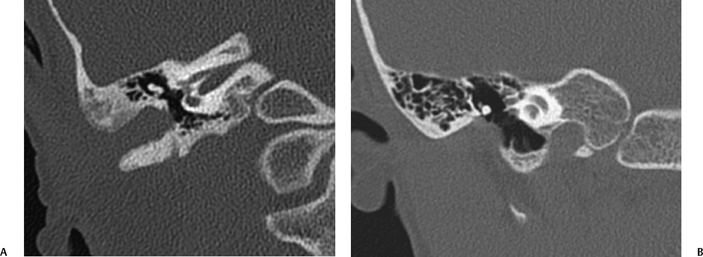
Fig. 2.5 (A) Coronal computed tomography scan shows right-sided membranous atresia (note absence of bony atresia plate). The malleus and incus are deformed and fused laterally. The middle ear cavity is slightly small. (B) On the left side, similar findings are seen. The deformed incus is laterally fused.
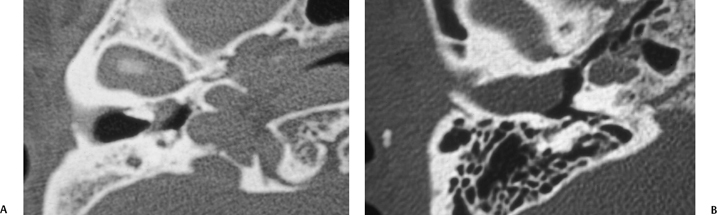
Fig. 2.6 (A) Axial computed tomography (CT) scan shows bone stenosis of the medial external auditory canal (EAC) with a small soft tissue mass medial, proven to be a cholesteatoma. (B) Axial CT in a different patient shows bone stenosis of the lateral aspect of the EAC with a large soft tissue mass medially, also proven to be a cholesteatoma.
Of the syndromes resulting in microtia and EAC atresia, Goldenhar syndrome or hemifacial microsomia is the most common and is also the second most common craniofacial anomaly after cleft lip and palate. There are four major components to Goldenhar syndrome: deformity of the auricle, EAC atresia, malformation of the tympanic cavity, and ossicular abnormalities causing conductive hearing loss. Occasionally, there is also sensorineural hearing loss due to stria vascularis and semicircular canal or cochlear abnormalities as well as hypoplastic or atretic oval window.21,22 Pfeiffer syndrome is another cause of EAC stenosis/atresia that results in moderate to severe conductive or mixed hearing loss in patients with cranio-facial abnormalities. In addition to the stenosis and/or atresia of the EAC, there is hypoplasia of the middle ear cavity and occasionally hypoplastic ossicles. Typically, the inner ear anatomy is normal, but middle ear effusions are frequently seen. Therefore, when Pfeiffer syndrome patients receive CT for craniofacial anomalies, examination of the temporal bones should also be performed.23
Treacher Collins syndrome is an autosomal dominant genetic disorder that is also associated with temporal bone abnormalities. Eighty-five percent of these patients present with bilateral microtia as well as bilateral abnormalities of the EAC, TM, ossicles, and middle ear cavity. These patients will also have midface hypoplasia and micrognathia.24,25
Pierre Robin syndrome (also known as Pierre Robin sequence) demonstrates retrognathia, glossoptosis, and bilateral cleft palate, as well as ear abnormalities. The ear abnormalities include an abnormal pinna and ossicles, as well as abnormal stapes footplates in 50% of patients. Aplasia of the lateral semicircular canals (LSCCs), large vestibular aqueducts, and unusually large otoconia are also seen, but not EAC atresia or stenosis.26 Because the middle ear arises from the branchial arches, whereas the inner ear does not, labyrinthine abnormalities such as aplasia and partitioning deficiencies are typically not associated with microtia and EAC atresia/stenosis.
When we interpret CT studies of the temporal bones performed in patients with congenital abnormalities, our report addresses the following:
- Extent of EAC atresia and its nature (membranous, bony, or a combination of both)
- Thickness of the atresia plate
- Amount of mastoid pneumatization
- Normal or abnormal middle cranial fossa level (especially if too low)
- Temporomandibular joint location
- Presence or absence of CH (Fig. 2.7)
- Ossicular fusion—incudomallear and incudostapedial joint maintenance, and if the ossicles are fused to the atresia plate (Fig. 2.8). (Rotation of the long incus process resulting in an obtuse angle to the lenticular process and the incudomallear and incudostapedial joints being visible on the same axial slice should also be mentioned.)
- Stapes present, dysplastic, or absent (may be useful information if a prosthesis placement is required)
- Size of the oval window (normal size is 2 mm in vertical diameter)27,28
- Size of the middle ear cavity in all three axes (if < 3 mm in width from the lateral margin to the cochlear promontory, surgery may be precluded)28
- Inner ear structures and size of the internal auditory canal (IAC; rarely affected with EAC dysplasia, but if abnormal may exclude surgical intervention. If the IAC is small, there can be associated cochlear nerve deficiency, but deficiency of the cochlear nerve can also be present with a normal-size IAC, and MRI may be indicated to assess the status of the cochlear nerve).29,30
- Course of the facial nerve with special attention to its horizontal portion in the middle ear and its descending portion (location of the stylomastoid foramen) (Fig. 2.9)
- Size of the oval window (normal size is 2 mm in vertical diameter)27,28
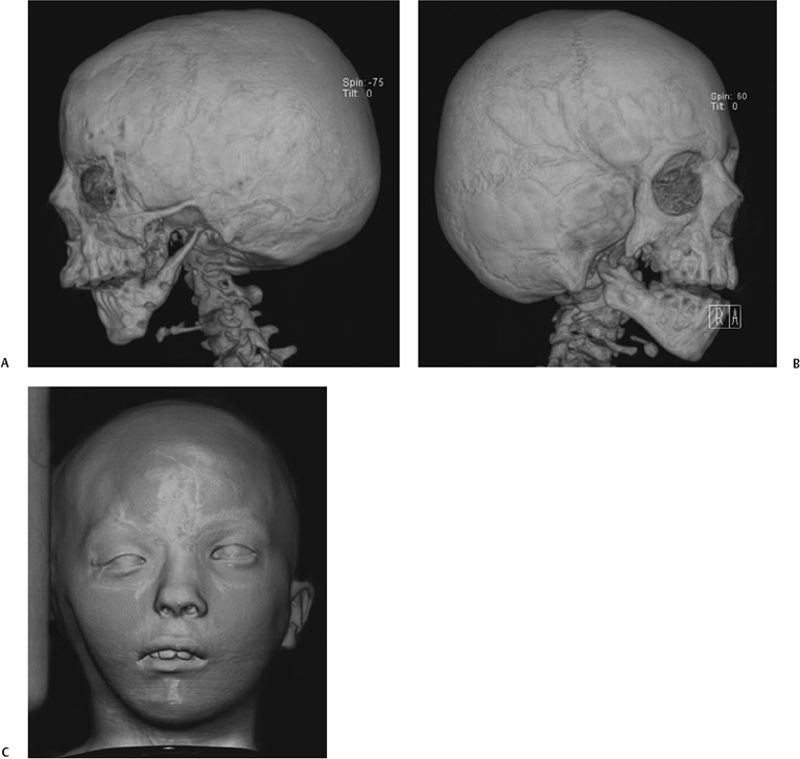
Fig. 2.7 (A) Three-dimensional computed tomography (CT) surface rendering in a patient with hemifacial microsomia shows an absent external auditory canal, a hypoplastic zygomatic arch, and a thin, small vertical mandibular ramus. (B) In a different patient also with hemifacial microsoma, CT surface rendering of bone shows an absent EAC, a partially absent zygomatic arch, and a hypoplastic ipsilateral mandible. (C) In the same patient, CT surface rendering of skin shows hypoplasia of the right side of the face and an absent pinna on that side. (See Color Plate Fig. 2.7.)
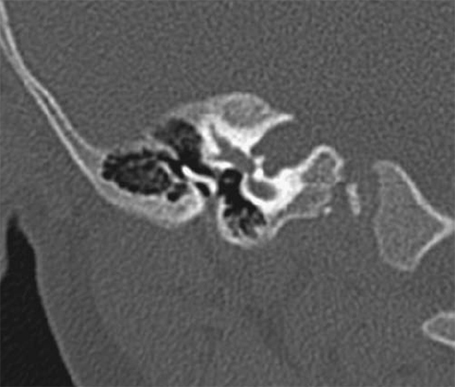
Fig. 2.8 Coronal computed tomography scan shows severe bony stenosis and membranous atresia. The small external auditory canal is vertically oriented. The malleus and incus are fused and deformed, and this ossicular mass is fused laterally. The facial nerve is directly behind the deformed ossicles, and the middle ear cavity is small.
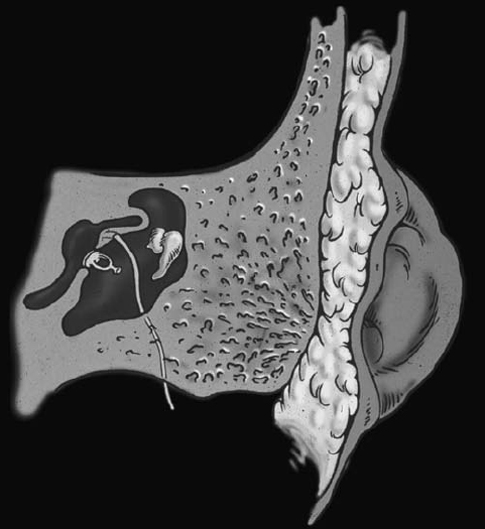
Fig. 2.9 Drawing showing microtia, bony atresia, and deformed ossicles. The facial nerve descends anteriorly under the lateral semicircular canal and in front to the stapes and thus is prone to injuries when drilling the atresia plate to gain access to the middle ear cavity. (Courtesy of Suresh Mukherji, MD, Ann Arbor, MI.) (See Color Plate Fig. 2.9.)
Surgical reconstruction for auricular and EAC atresia are considered separately. Usually, microtia is a cosmetic procedure that is undertaken prior to entering grade school. A variety of approaches and materials have been used, but currently autogenous rib grafting using a multistage operation produces excellent results when performed by a highly experienced surgeon. Others have used alloplastic materials with very good results as well. The creation of an EAC with an intact conductive hearing mechanism usually follows the auricular surgery so there is no interference with the healing of the skin flaps and the implanted framework. EAC reconstruction is undertaken either to improve hearing or when CH is present. Thus, bilateral involvement represents a clear indication for surgical intervention. Following early identification, these children are fit with a bone conduction hearing aid to ensure normal auditory receptive abilities prior to surgical intervention. With this device, speech and language acquisition usually proceeds normally. Atresiaplasty in unilateral cases is reserved for those patients where the anatomy is favorable, the family expectations are appropriate, and the child is very cooperative. Favorable anatomy is generally present when inner ear and facial nerve morphology is normal, temporal bone pneumatization is good, and the ossicular chain is only mildly deformed. Jahrsdorfer et al24 has created a classification system to help in decision making in this regard.
For atresiaplasty, the EAC is created by drilling posterior to the glenoid fossa and anterior to the mastoid air cells, entering the epitympanum and superior aspect of the middle ear space just below the level of the middle cranial fossa dura.28 The ossicular chain is mobilized from the atresia plate, and a TM is created using a temporalis fascia graft. Finally, the canal is lined with a split-thickness skin graft, and a meatoplasty is created by attaching the skin graft to a newly created meatal opening.14 Good hearing results (air–bone gap 25 dB) can usually be obtained in 75% of patients and are dependent on favorable anatomical factors. In cases where good hearing is not realized from the procedure, a conventional hearing aid can be used with excellent results. For children where anatomical factors preclude EAC reconstruction or the risks seem unacceptable to the family, an osseointegrated cochlear stimulator (BAHA device, Cochlear Corp., Englewood, CO) can be considered.
Postoperative complications from EAC atresia reconstruction include facial paralysis, sensorineural hearing loss, conductive hearing loss, TM perforation, cerebrospinal fluid (CSF) leak, and meatal stenosis secondary to bony regrowth or soft tissue stenosis. This latter complication is widely considered the most frequently occurring.31 The most dreaded complication of EAC atresia surgery is facial nerve injury. Although rare, the facial nerve is at risk of injury in both its descending segment and in the extracranial portion. In the intratemporal portion, anterior displacement of the descending segment is the norm, making it most vulnerable while drilling the ear canal inferiorly. The extratemporal portion of the facial nerve is at risk of injury during meatoplasty, especially when the cartilaginous framework from the microtia repair lies anterior to the newly created ear canal, requiring mobilization for realignment. Other complications of atresiaplasty include persistent conductive hearing loss from ear canal stenosis, TM perforation or lateralization, and ossicular chain discontinuity or fixation. Sensorineural hearing loss can occur from inadvertent labyrinthine injury while drilling around the ossicular chain. We have recently started to use a laser to lyse the final attachments of the ossicular chain to avoid such drill-induced trauma.30
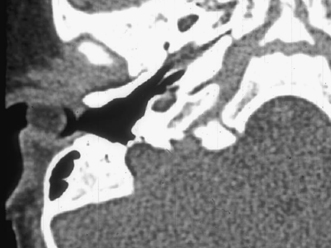
Fig. 2.10 Axial computed tomography (CT) scan shows a cystic rounded mass in the lateral external auditory canal (EAC). This is a proven type 1 branchial cleft cyst, which extended into the EAC via a defect in its floor.
Brachial Cleft Cyst
A brachial cleft cyst (BCC) is a congenital lesion that can occur in the area of the EAC, and is the cause of one third of pediatric nonmalignant lesions in the region of the parotid gland requiring surgery.32 BCCs arise from lack of involution of structures from the first through fourth branchial arches and are classified by location. Pathologically, BCCs are composed of a thin fibrous pseudocapsule with central squamoid epithelium and occasionally lymphocytic and germinal tissues. The majority of BCCs are simple cysts (two thirds) with thick mucous contents, no cutaneous or airway opening, and 3 cm or less in size. Due to their histologic components, BCCs are similar in their imaging appearance, regardless of their location. Rarely, a BCC undergoes malignant transformation into squamous cell carcinoma.33 On physical examination, a BCC is compressible due to its fluid components and usually painless. These lesions have a tendency to enlarge with upper respiratory tract infections due to lymphoid secretions from follicles in the wall of the cyst and may become painful even in the absence of infection.
BCCs are readily evaluated with CT. Their appearance is that of a well-circumscribed mass with fluidlike components centrally (Fig. 2.10).34 On MRI, a BCC is of low T1- and high T2-weighted signal and may exhibit rim enhancement following contrast administration. Increased proteinaceous contents may lead to higher T1-weighted signal and occasionally low T2-weighted signal intensity. In the absence of infection, FLAIR imaging will demonstrate the central fluid to have low signal. If there is accompanying infection, surrounding soft tissue edema (stranding of fat) will be present on CT and MRI, and the rim has a tendency to be thicker, nodular, and to enhance. MRI may be useful for locating a sinus/fistula. CT better assesses bony abnormalities.35
Fistulography can also be performed to better assess the course, anatomy, and topography of a fistulous tract, which helps improve the rates of complete resection of a fistula associated with a BCC.36
In this chapter, we will concentrate on first BCCs because these occur in the region of the EAC. First BCCs are less common than second BCC cysts and account for less than 8% of all BCCs. A first BCC arises in a periauricular location, often parallels the EAC in orientation, and can be enveloped by the parotid gland. A congenital tract or sinus from the BCC may communicate with the EAC. Due to this embryological development, a first BCC can be associated with other first cleft anomalies, including heterotopic salivary gland tissue, which has a predisposition for malignant transformation,37 or CH.38 Although there are two possible subtypes of a first BCC, there are no strict or well-defined histological and anatomical features for each one. The probability of an associated sinus with a first BCC is 56%, and a fistulous tract to the EAC at the cartilaginous–bony junction occurs in 31%, with the remaining first BCCs having a simply cystic appearance.39 A first BBC usually presents while the patient is a child, one third of patients presenting acutely due to infection and two thirds presenting with asymptomatic swelling, but may also present in teenagers or young adults if the lesion is purely cystic and not infected.40 On rare occasions with any BCC, there can also be communication with the skin surface, producing a fistula.
Commonly, a BCC is treated by complete resection to prevent recurrence and reduce complications.41 Because many BCCs present as an abscess, they can be improperly diagnosed and treated with simple incision and drainage rather than complete excision.42 The surgeon must be careful to not injure the adjacent extratemporal portion of the seventh cranial nerve.39,43 Due to variable relationships of BCCs to the facial nerve, imaging is important for surgical planning. Even with identification of the facial nerve at surgery, there is a 21% incidence of temporary and 1% incidence of permanent facial palsy after resection of a first BCC. Additionally, the probability of facial nerve injury increases with the number of previous infections and surgeries.44 More recently, methods other than surgical excision have been described for treatment of a BCC. An endoscopic approach for excision of a BCC through a small transcervical incision has been described and is said to have minimal morbidity.45 However, data regarding the safety and efficacy of this approach are lacking. Also described is the use of ethanol injections for sclerotherapy of a BCC.46
The differential diagnosis for a BCC includes other cystic lesions, such as lymphangiomas, which are usually multiloculated, nonenhancing, and transspatial lesions. Cystic lesions in the region of the parotid tail include lymphoepithelial cysts with AIDS, cysts of the parotid in Sjögren’s syndrome, and less commonly, cysts in Reiter’s syndrome. Also, an infected intraparotid or suppurative jugular lymph node, or rarely, an intranodal necrotic metastasis from squamous cell or thyroid cancers must be considered.47,48 Lymphoma, both Hodgkin and non-Hodgkin types, are also in the differential diagnosis, but typically demonstrate multiple masses/enlarged nodes.49
Another uncommon entity in the differential is a cystic schwannoma from the facial nerve.50
Cystic Hygroma and Cystic Lymphangioma
Lymphangiomas occur less frequently than BCCs. Embryologically, lymphangiomas may have two possible origins: they may arise secondary to failure of embryonic fusion between the central venous system and lymph sacs or from sequestration of lymph sacs. In either case, the condition is associated with Turner’s and Noonan’s syndromes as well as with fetal alcohol syndrome. Clinically, most cystic hygromas and lymphangiomas present in the first 2 years of life and only rarely in adulthood.51 On physical examination, lymphangiomas are compressible masses, usually involving the submandibular and posterior triangle regions, which may result in airway obstruction. Lymphangiomas may also extend to the mucosal surfaces of the oropharynx, tongue, or airway. Pathologically, they are composed of endothelial-lined dilated endolymphatic spaces with septations of variable thickness and at times vascular structures. Depending upon the size of the lymphatic spaces within a lesion, there is a pathologic continuum with cavernous hemangioma, capillary lymphangiomas (least common form with the smallest spaces), and vasculolymphatic malformations.
Different lesion types cannot always be differentiated by imaging, but a cystic hygroma is the most common type.52
Contrast administration is needed to assess these lesions because the presence of venous components may change the surgical approach. On imaging, the most distinguishing feature of a cystic hygroma or lymphangioma is a tendency to insinuate itself into multiple compartments in a transspatial manner and to surround normal anatomical structures such as muscles and blood vessels. Hence, a lymphangioma is less well circumscribed than a BCC. Lymphangiomas are most commonly multilocular and nonenhancing, although they may rarely enhance if there is superimposed infection. Additionally, when infection is present, it may extend outside the lesion.53 On CT, these lesions may display fluid-fluid levels and occasionally show venous (enhancing) components or areas of enhancing soft tissue. On MRI, the lesions may have a simple cystic appearance, but if there has been prior hemorrhage or proteinaceous fluid, they will have high T1-weighted signal and/or fluid-fluid levels.
The differential diagnosis for a lymphangioma includes other slowly growing neck masses, including neurofibroma, schwannoma, hemangioma, vascular malformation, and sublingual salivary mucocele (i.e., ranula or pseudocyst). Acute swelling and a more rapid presentation is seen in suppurative lymphadenopathy secondary to sinusitis, odontogenic infection, or abscess. A BCC with superimposed infection is more common than a lymphangioma. If there has been a rapid clinical change such as swelling and/or cranial nerve deficits, a malignant process such as rhabdomyosarcoma, Langerhans cell histiocytosis, Ewing sarcoma, osteogenic sarcoma, or metastatic neuroblastoma should be considered.54
Lymphangiomas can be assessed before surgery with ultrasound (US), CT, and/or MRI. In our institution, the majority of patients are assessed with contrast-enhanced neck CT because the diagnosis is not always known at presentation, and CT can exclude most of the other lesions mentioned above in the differential. Imaging prior to surgical intervention allows for staging of the lesion, determination of unilaterality or bilaterality, infra- or suprahyoid extent, and mediastinal involvement. These differences in staging predict surgical outcomes as well as complication and morbidity rates.55 MRI reveals associated pathology not seen with US in 20% of patients.56 MRI is also useful for assessment of the amount of tracheal/airway compromise.57
Standard treatment of lymphangiomas is surgical. Recently, new types of intervention are being employed, including US-guided cavity aspiration with injection of bleomycin, which has resulted in a good response in more than 50% of patients.58,59 Also, administration of a four-dose injection regimen of OK-432 (Picibanil) at 6- to 8-week intervals has shown an 86% success rate for obliteration or substantial reduction (> 60%) of the cystic hygromas.60 In a minority of patients, lymphangiomas may resolve/involute on their own.61 Lymphangiomas are known to have a tendency to recur, particularly if nonencapsulated; therefore, they need to be followed closely regardless of the course of therapy utilized.
Foramen of Huschke
Approximately 4.6% of patients have an area of medial bony dehiscence involving the anterior and inferior EAC called the foramen of Huschke. Normally, the foramen of Huschke obliterates during childhood or infancy as the U-shaped EAC cartilage undergoes fusion into a complete ring. On physical examination, a persistent foramen may present as a small polyp or punctum on the anterior wall of the EAC. These congenital fistulas are rare. The foramen is readily detected with high-resolution CT, is located posterior and medial to the temporomandibular joint (TMJ), and measures ∼3 to 4 mm in size. A patent foramen of Huschke is more common in females and can cause transient otorrhea from TMJ synovial fluid. Rarely, soft tissue posterior to the TMJ meniscus can herniate into the EAC during mouth closure. Rarely, this tract can act as a portal for the spread of infection or tumor between the EAC and TMJ. Its presence can be identified with high-resolution (0.6 mm thick) CT imaging, is seen as bony EAC thinning (< 1.0 mm) anteriorly and inferiorly, and is usually bilateral. Normally, the foramen of Huschke closes by the age of 5 years, and its persistence is an anatomical variant.62
Salivary otorrhea from a patent foramen of Huschke presents with serous discharge from the EAC that occurs more frequently with meals. This fluid demonstrates the presence of amylase when counterstained with iodine on a starch agar plate. Sialography may show the presence of a fistula to the EAC or just chronic parotid sialadenitis without a definite fistula. MRI reveals bright T2-weighted signal within the adjacent parotid secondary to the sialadenitis and/or fluid in the EAC. Usually, the defect is repaired surgically using temporalis fascia and tragal perichondrial grafts.63
Inflammatory
Otitis Externa
There are six forms of otitis externa (OE; also known as external otitis); acute, chronic, eczematous (dermatitis, psoriasis, lupus, or infantile eczema related), fungal, and necrotizing/malignant.
Acute Uncomplicated External Otitis
Acute uncomplicated external otitis or swimmer’s ear is the most common external ear infection. Patients with OE present with pain, erythema, swelling, and severe tragal and auricular motion tenderness. Occasionally, a conductive hearing loss may be present when canal swelling has obliterated the patency of the EAC.64,65 Most commonly, the condition is caused by a bacterial infection from Pseudomonas aeruginosa or Staphylococcus aureus. Otomycosis or fungal OE usually results from infection from either Candida or Aspergillus species. Presentation usually follows the administration of multiple oral or topical antibacterial medications and can be quite refractory to conventional therapy.66 There is also an increased risk of otomycosis among diabetic and/or immunocompromised patients.67 When otomycosis complicates perforated otitis media or otitis media with a tympanostomy tube, therapy can be very challenging because most medications that are active against fungal species have not been approved by the U.S. Food and Drug Administration or are safe for middle ear usage. Uncommonly, acute OE can result from viral infection. Ramsay Hunt syndrome or herpes zoster oticus is due to infection of the seventh and eighth cranial nerves by reactivation of latent herpes zoster virus in the geniculate, spiral (i.e., cochlear), and/or Scarpa’s (i.e., vestibular) ganglia. Classically, this disease presents with acute facial palsy and a vesicular eruption in the distribution of the somatosensory fibers of the facial nerve within the EAC and auricle.68 When the vestibulocochlear nerve is involved, sensorineural hearing loss and vertigo may be present to varying degrees. Treatment of this disorder usually requires systemic corticosteroid therapy and an antiviral medication with efficacy against the offending agent (i.e., valacyclovir).
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


