5 The Inner Ear and Otodystrophies
Phylogeny and Embryology
The phylogeny of the human ear has been the subject of considerable study. In most aquatic organisms, there is only a water motion detection system.1 The initial evolution into what we understand as the “ear” begins with the development of structures analogous to the phylogenetically older utricle and semicircular canals (SCC; posterior and superior only). Later vertebrates evolved a third (lateral) semicircular canal (LSCC) and a saccule. A primitive cochlear duct, the lagena, further evolves as an outpouching of the saccule. The major alteration in the evolutionary ladder occurs when these structures become isolated from the external environment and endolymph replaces seawater as the essential fluid. This occurs simultaneously with the formation of a distinct tympanomastoid compartment, which itself represents the functional adaptation of the aquatic gill; the ossicular chain probably develops as seawater is no longer available at this stage to serve as a conductive medium.2
A simplification of the embryological development of the ear is a difficult task. The reader is referred to other textbooks for more detailed explanations of this process.3 Suffice it to say that the maturation of the inner ear has three main phases: (1) development (3rd to 11th week), (2) growth (11th to 16th week), and (3) ossification (16th to 24th week). The development of the sensory epithelium within the membranous labyrinth occurs simultaneously with growth and ossification (8th to 24th week) (Table 5.1).
The inner ear arises from the otic placode, which is a thickening of the surface neuroectoderm located between the first branchial groove and the hindbrain. This process occurs early in the third gestational week. Each otic placode invaginates and sinks below the surface neuroectoderm into the underlying mesenchyme. This results in formation of the otic pit. The edges of the otic pit fuse to form the otic vesicle (otocyst).4 The otic vesicle is the precursor of the membranous labyrinth.
The otic vesicle divides into a dorsal utricular portion, which forms the utricle, SCCs, and endolymphatic duct (ELD), as well as a ventral saccular portion that gives rise to the saccule and cochlear duct. The cochlear duct completes one turn by the 8th week, and two turns by the 9th to 10th week. By the end of the 10th week, the entire membranous labyrinth is identifiable and formation of the 2 1/2 to 2 3/4 cochlear turns is complete. The fetus is able to hear with maturation of the organ of Corti, which occurs by the 22nd to 24th week of gestation.
The otic capsule develops as a cartilaginous condensation of mesenchyme around the otic vesicle between the fourth and eighth weeks of gestation. Growth of the otic capsule continues to the 16th gestational week. During the growth of the otic capsule, vacuoles begin to arise in the developing cartilaginous otic capsule. These vacuoles eventually coalesce to form the perilymphatic space. The newly created perilymphatic space contains perilymph fluid, which surrounds and bathes the membranous labyrinth. Ossification of the otic capsule occurs between weeks 16 and 24 via 14 ossification centers. The end result is the bony labyrinth.
Otic capsule ossification is unique in that (1) there are a large number of ossification centers—14—considering the small size of the final product; (2) there is no epiphy-seal growth (centers fused directly); and (3) maturation is arrested in a primary state of ossification (i.e., endochondral bone persists).
The membranous part of the SCC system begins to develop as two fingerlike extensions from the utricular side of the otic vesicle at about the fourth week of gestation.5
| Primordium | Week Complete |
| Otic placode | 3rd |
| Otic pit | 4th |
| Otic vesicle (otocyst) | 5th |
| Ventral pouch of otocyst: | |
| Cochlear duct | 8th |
| Saccule | 11th |
| Dorsal pouch of otocyst: | |
| Endolymphatic duct | 11th |
| Utricle and semicircular ducts | 11th |
| Semicircular canals | 19th-22nd |
| Labyrinthine ossification | 23rd |
| Total development | 26th |
We have presented an overview of the development of the inner ear. A more detailed discussion of specific structures will be included in the following sections.
Anatomy and Function
Overview
The inner ear consists of a membranous (endolymphatic) labyrinth containing the functional sensory epithelium surrounded by an osseous (bony) labyrinth with an interposed perilymphatic labyrinth (Fig. 5.1A, Fig. 5.1B; see color plates) (Table 5.2).
The membranous labyrinth consists of the utricle, saccule, semicircular ducts, cochlear duct (scala media), and endolymphatic duct and sac. All of these structures contain endolymph, the functional fluid of the inner ear that bathes and nourishes the sensory epithelium. Endolymph is rich in potassium and low in sodium, similar to intracellular fluid (Fig. 5.1B).3,4,6–8
The osseous labyrinth is the bony shell that surrounds the membranous labyrinth. The utricle and saccule (membranous labyrinth) are housed within the vestibule (bony labyrinth), the semicircular ducts (membranous labyrinth) within the semicircular canals (bony labyrinth), and the ELD (membranous labyrinth) within the proximal vestibular aqueduct (bony labyrinth) (Fig. 5.1A).
The perilymphatic labyrinth is interposed between the membranous and bony labyrinth. Perilymph is rich in sodium and low in potassium. This composition is similar to extracellular and cerebrospinal fluid. Perilymph is located within the vestibule surrounding the utricle and saccule, between the semicircular ducts and walls of the SCCs, and within the vestibular aqueduct surrounding the ELD.4,9–11 The scala vestibuli and scala tympani are perilymph-containing structures within the cochlea that parallel the endolymph-containing cochlear duct (scala media) and are crucial to the sound conduction mechanism (Fig. 5.1C and Fig. 5.1D; see color plates).
Perilymph is also the exclusive fluid content of the cochlear aqueduct (CA).
The Vestibule and Semicircular Canals
Anatomy
The vestibule is the largest cavity of the bony labyrinth, measuring 4 to 6 mm at maximal diameter. The medial wall of the vestibule is unique in that it contains two distinct depressions—the elliptical recess posterosuperiorly and the spherical recess anteroinferiorly (Fig. 5.1A, Fig. 5.2B, Fig. 5.3B, and Fig. 5.4C).3,10,11 Between these two concavities lies the vestibular crest, a ridge that divides posteriorly into two limbs bounding an additional depression, the cochlear recess, which is the most proximal portion of the cochlear apparatus leading to the scala vestibuli (cochlea). These structures are not typically seen on routine imaging as of this writing.
The utricle is firmly anchored within the elliptical recess by connective tissue and filaments of the utricular branch of the superior vestibular nerve.12 Similarly, the saccule is adherent to the spherical recess by fibrous tissue and the saccular branch of the inferior vestibular nerve.
The utricle and saccule communicate with each other via the utriculosaccular duct. The utricle also communicates with the endolymphatic sinus (as well as the SCCs) via the utricular duct. In addition to the communications with the utricle, the saccule communicates with the endolymphatic sinus (via the saccular duct) and the cochlea (via the ductus reuniens).3,4,6,13,14
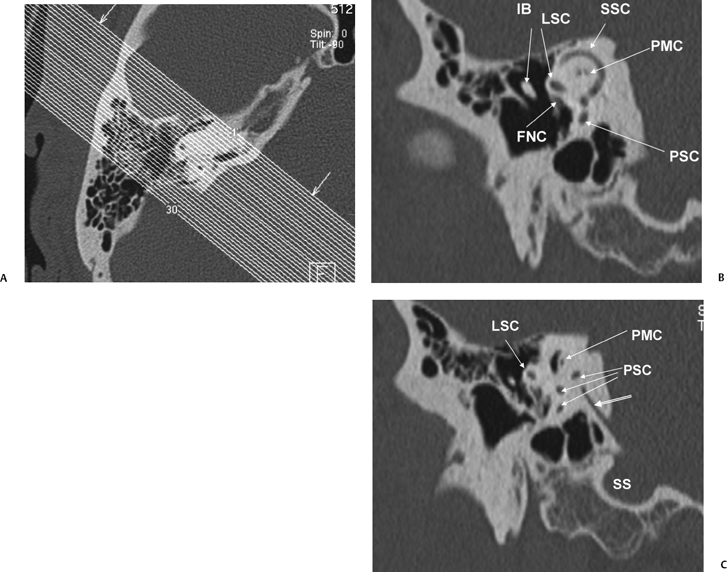
On the posterior wall of the utricle reside five openings for the three semicircular ducts. Remember that when we use the term ducts, we are referring to the functioning endolymph containing apparatus, and when we use the term canals, we are referring to the bony covering only, which is separated from the ducts by perilymph. There are five openings instead of six because the superior and posterior semicircular ducts/canals have a common crus (Fig. 5.3C, Fig. 5.4C, Fig. 5.4E, and Fig. 5.4F). The osseous superior border of the SSCC is responsible for a ridge along the roof of the petrous bone known as the arcuate eminence (Fig. 5.3D, Fig. 5.5A, and Fig. 5.5B). Bony dehiscence at this site may result in significant clinical symptomatology and is discussed in detail later in the chapter. The LSCC is 30 degrees off the horizontal plane and provides much of the anatomic medial wall of the epitympanum (attic) and aditus ad antrum (Fig. 5.2B, Fig. 5.2C, Fig. 5.3B, Fig. 5.3C, and Fig. 5.3D). Each of the semicircular ducts/canals is orthogonal with one another. The superior and lateral canals/ducts are innervated by the superior vestibular nerve, the posterior SCC/duct by the inferior vestibular nerve (singular nerve).
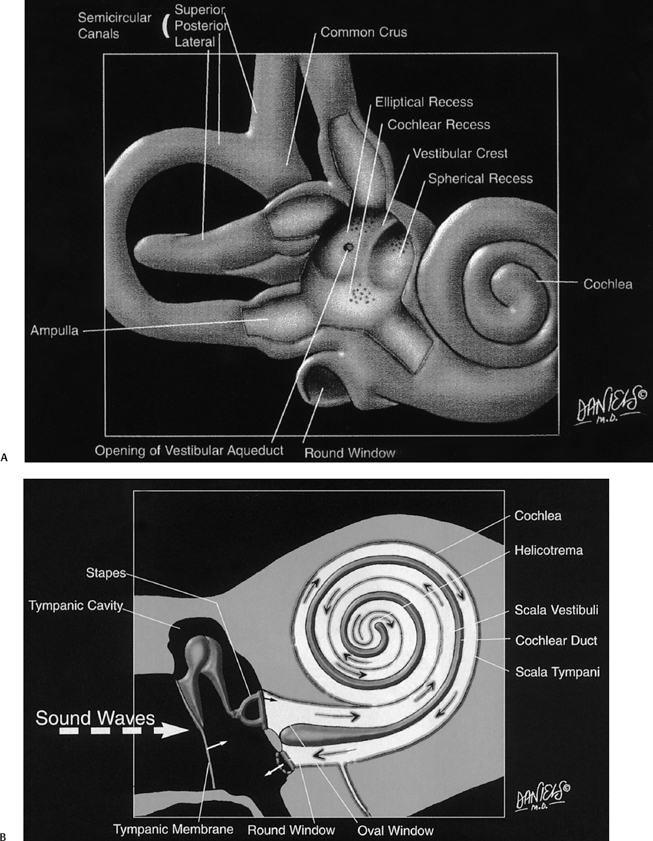
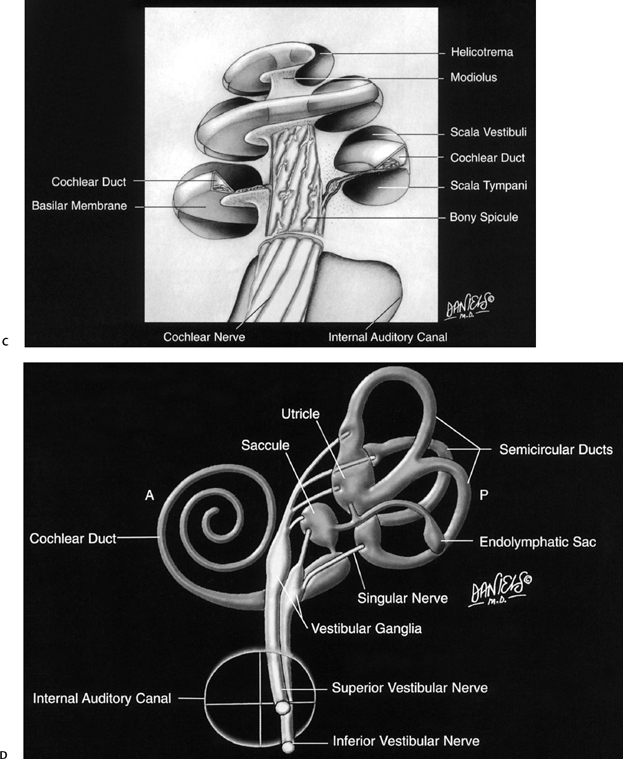
Fig. 5.1 Artist renderings. Normal inner ear anatomy. (A) Bony labyrinth with emphasis on structure of posterior wall of vestibule (Color Plate 5.1A). (B) Membranous labyrinth and subtended neural structures (Color Plate 5.1B). Artist renderings. Normal inner ear anatomy. (C) Cochlea (Color Plate 5.1C). (D) Movement of sound waves within the cochlea. See text and Color Plate 5.1D. ([A–D] Courtesy of David L. Daniels, MD, with permission.)
| Bony Labyrinth | Membranous Labyrinth (Endolymphatic) |
| Cochlea | Cochlear duct |
| Vestibule | Utricle and saccule |
| Vestibular aqueduct | Endolymphatic duct/sac |
| Semicircular canals | Semicircular ducts |
Function
The utricle, saccule, and semicircular ducts are involved with balance and maintenance of a stable retinal image. The utricle and saccule are collectively referred to as the static labyrinth as their function is to detect the position of the head relative to gravity. They each have a focal concentration of sensory receptors (maculae) located at right angles to each other and consisting of ciliated hair cells and tiny crystals of calcium carbonate (otoliths) embedded in a gelatinous mass. These otoliths respond to gravitational pull; therefore, changes in head position distort and stimulate the hair cells.
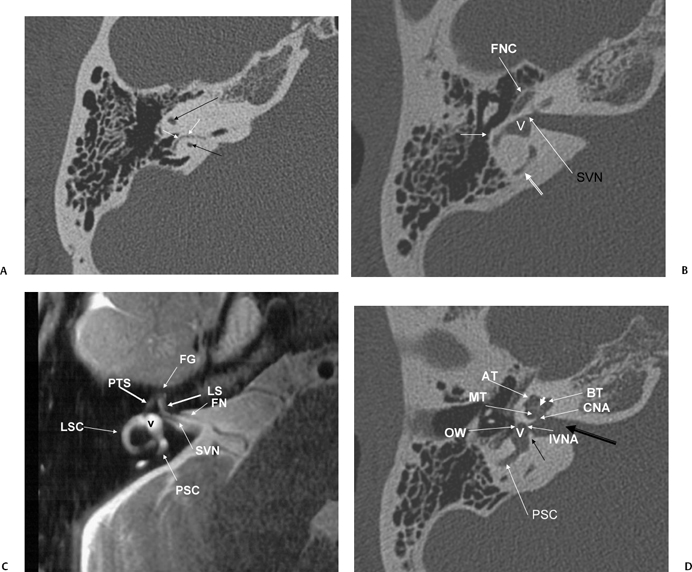
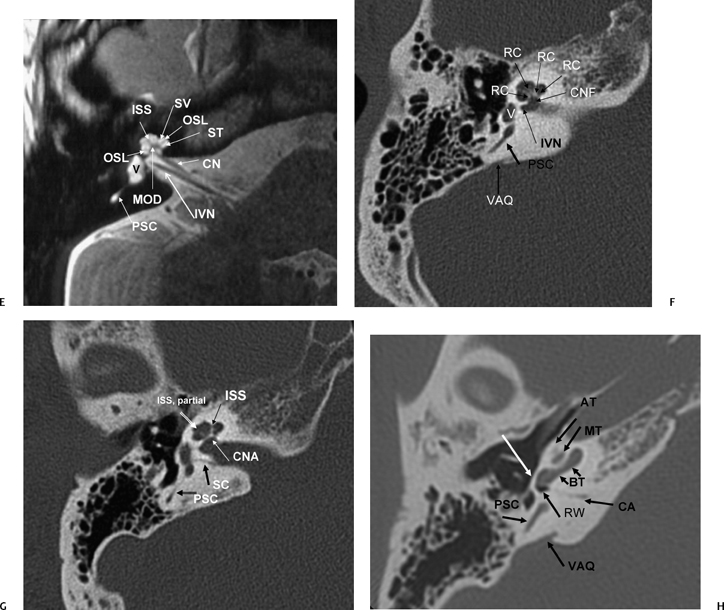
Axial T2WI, right ear (CN, cochlear nerve; IVN, inferior vestibular nerve; OSL, osseous spiral lamina; PSC, posterior semicircular canal; V, vestibule) contains the cochlear duct (scala media) and separates the scala vestibuli (SV) and scala tympani (ST). The interscalar septum (ISS) separates the apical turn from the middle turn. Note that the modiolus (MOD) has a classic “trapezoid” shape. (F) Axial CT (IVN, canal for inferior vestibular nerve; V, vestibule; CNF, cochlear neural foramen; VAQ, vestibular aqueduct; RC, canal of Rosenthal [through which cochlear nerve fibers travel through the osseous spiral lamina from the modiolus]). (G) Axial CT (PSC, posterior semicircular canal; SC, singular canal, singular nerve to posterior semicircular canal; CNA, cochlear nerve aperture; ISS, medial interscalar septum; ISS, partial, lateral interscalar septum [This is a normal variation compatible with normal cochlear function and should not be confused with pathologic modiolar deficiency.]). (H) Axial CT (AT, apical turn of cochlea; MT, middle turn of the cochlea; BT, basilar turn of the cochlea; RW, round window; VAQ, vestibular aqueduct [and adjacent fovea harboring endolymphatic sac]; PSC, posterior semicircular canal; white arrow, promontory; CA, cochlear aqueduct [otic capsule segment]).
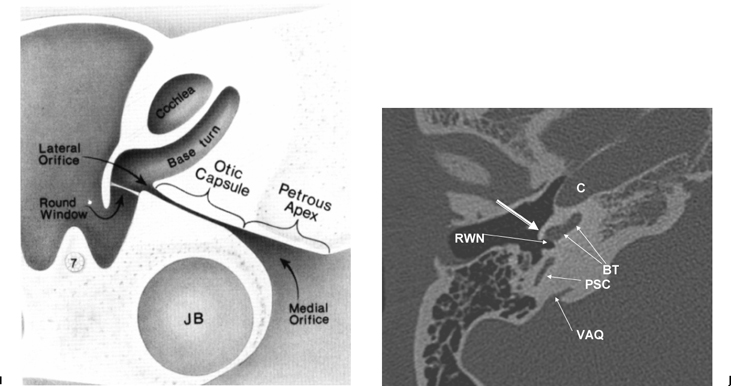
(I) Drawing emphasizing cochlear aqueduct. (From Jackler RK, Hwang PH. Enlargement of the cochlear aqueduct: fact or fiction? Otolaryngol Head Neck Surg 1993;109:14–25. Used with permission.) (J) Axial CT (PSC, posterior semicircular canal; BT, basilar turn of the cochlea; VAQ, vestibular aqueduct; C, carotid canal, horizontal portion; RWN, round window niche) promontory [bony covering of basilar turn of the cochlea] (outlined white arrow).
The semicircular ducts are collectively referred to as the kinetic labyrinth because they respond to rotational or angular acceleration. Each of the semicircular ducts subtends two thirds of a circle and contains an ampulla for transmission of vestibular nerve fibers. These ampullae are analogous to the maculae of the utricle and saccule in that they contain hair cells. Endolymph flow in response to angular head movements stimulates these hair cells. Each semicircular duct responds to a different rotational axis.
Vascular Supply
The arterial supply to the vestibule and semicircular canals/ducts arises from the labyrinthine branch (LB) of the anteroinferior cerebellar artery (AICA), which, in turn arises from the basilar artery at the junction between the proximal one third and distal two thirds of this vessel. On occasion, the AICA arises from the posteroinferior cerebellar artery (PICA) branch of the vertebral artery as an AICA–PICA trunk. The posterior, superior, and lateral semicircular canals/ducts are supplied via the anterior vestibular branch of the LB; the capsule and posterior semicircular canals/ducts receive nutrients via the posterior vestibular branch.
The petromastoid canal (PMC) is an anteriorly convex conduit usually measuring 0.5 to 1.0 mm passing between the SSCC and LSCC connecting the cranial cavity to the mastoid antrum, which should not be confused with fracture (Fig. 5.2A). There is an increase in the length of the PMC with increasing volume of perilabyrinthine pneumatization. It contains the subarcuate artery (branch of the AICA) and has the potential to transmit suppurative debris in either direction (similar to CA). These vessels supply the otic capsule of the semicircular canals as well as part of the vestibule, facial canal, and mastoid mucosa. The subarcuate fossa (SAF) is the medial orifice of the PMC. A wide PMC (> 2 mm) is not uncommon in children under 5 years of age (Fig. 5.6) and contains invaginated dura, which is a potential surgical hazard, particularly in cochlear implant (CI) candidates and those with chronic otitis media (COM).15
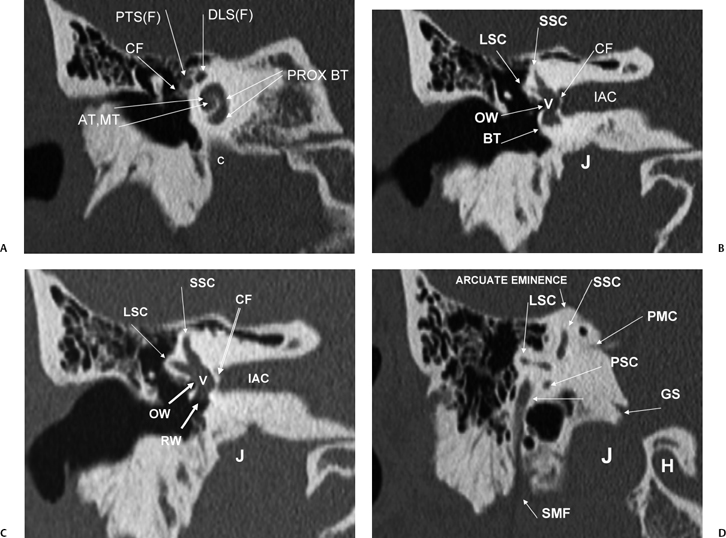
Fig. 5.3 Coronal computed tomography images. (A) AT, apical cochlear turn; MT, middle cochlear turn; PROX BT, proximal basilar turn; PTS(F), proximal tympanic segment, facial nerve canal; DLS(F), distal labyrinthine segment, facial nerve canal; CF, cochleariform process (insertion of tensor tympani tendon); c, carotid canal. (B) LSC, lateral semicircular canal; SSC, superior semicircular canal; OW, oval window; BT, distal basilar turn of the cochlea; IAC, internal auditory canal; CF, crista falciformis; J, jugular foramen. (C) LSC, lateral semicircular canal; SSC, superior semicircular canal; OW, oval window; IAC, internal auditory canal; RW, round window; IAC, internal auditory canal; CF, crista falciformis; J, jugular foramen; V, vestibule. (D) LSC, lateral semicircular canal; SSC, superior semicircular canal; PSC, posterior semicircular canal; PMC, petromastoid canal; GS, glossopharyngeal sulcus; SMF, stylomastoid foramen; J, jugular foramen; H, hypoglossal canal.
Innervation
Impulses from the utricular macula and the ampullae of the superior and lateral semicircular ducts travel via the superior vestibular nerve in the posterosuperior quadrant of the internal auditory canal (IAC). Impulses from the saccular macula and the ampulla of the posterior semicircular ducts travel primarily via the inferior vestibular nerve within the posteroinferior quadrant of the IAC. The saccule is actually innervated to some extent by all three segments of the vestibulocochlear nerve. The nerve of the posterior semicircular canal/duct, also referred to as the singular nerve, often travels separately from the inferior vestibular nerve, either within this quadrant or within an entire separate semicanal. This singular canal is clearly visualized on both coronal and axial computed tomography (CT) images. The aforementioned anatomic arrangement allows for surgical denervation of the posterior semicircular canal/duct (singular neurectomy), a procedure that has been used to treat patients with chronic benign positional vertigo (cupulolithiasis).3
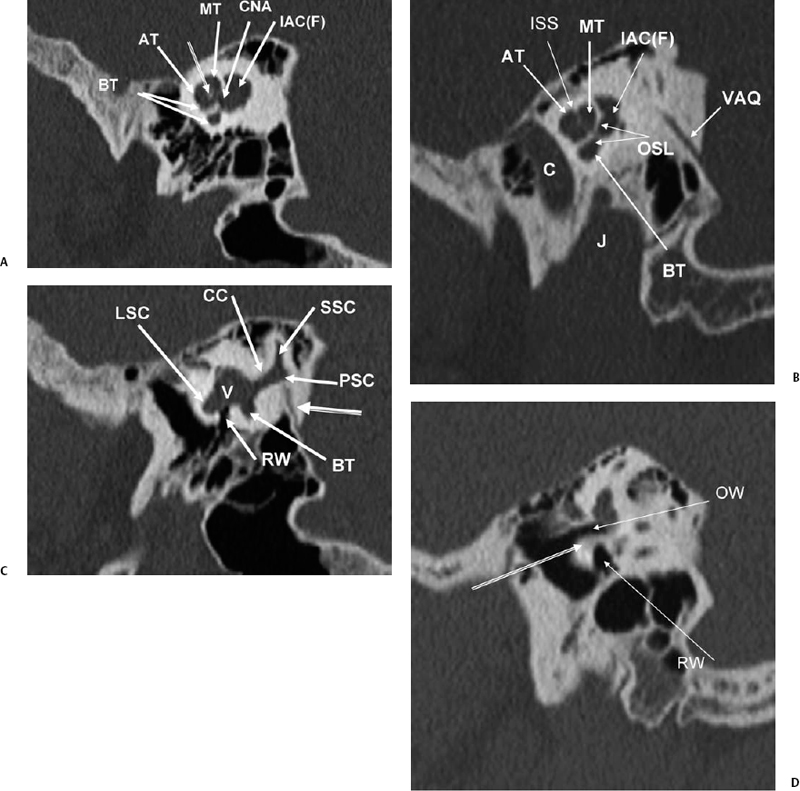
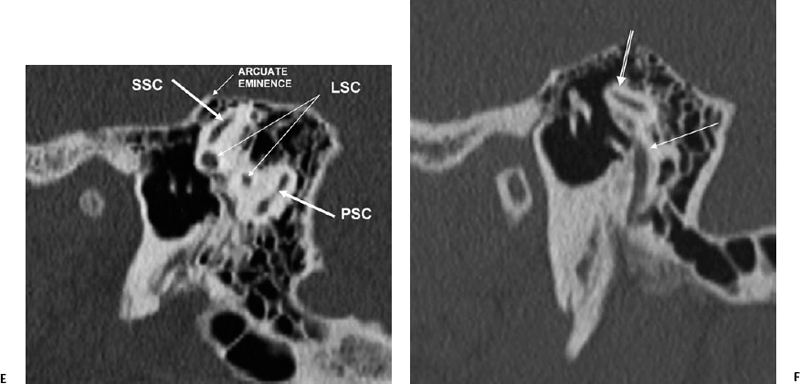
Fig. 5.4 Sagittal computed tomography images. (A) AT, anterior turn of the cochlea; BT, basilar turn of cochlea; MT, middle turn of the cochlea; CNA, cochlear nerve aperture; IAC(F), fundus of internal auditory canal. Modiolus (outlined arrow). (B) Apical (AT), middle (MT), and basilar (BT) turns of the cochlea; VAQ, vestibular aqueduct; IAC(F), fundus of internal auditory canal; OSL, osseous spiral lamina; ISS, interscalar septum; C, carotid canal; J, jugular foramen. (C) RW, round window (leading to scala tympani of basilar turn of the cochlea); LSC, lateral semicircular canal; SSC, superior semicircular canal; BT, basilar turn of the cochlea; CC, common crus (posterior and superior semicircular canals); V, vestibule. Vestibular aqueduct (arising from common crus) (outlined arrow). (D) Promontory (bony covering of basilar turn of the cochlea) (outlined arrow). OW, oval window; RW, round window. (E) LSC, lateral semicircular canal; PSC, posterior semicircular canal; SSC, superior semicircular canal. (F) Lateral semicircular canal (outlined arrow), mastoid segment, facial nerve canal (straight white arrow).
The vestibular (Scarpa’s) ganglion is situated within the IAC and is responsible for mediation of afferent and efferent impulses of the saccule, utricle, and all three semicircular ducts. As such, the vestibular nerve has five terminal branches. Four vestibular nuclei are located in the brainstem at the pontomedullary junction medial to the cochlear nuclei at the level of the floor of the fourth ventricle. Fibers of the vestibular nerve synapse in these nuclei. Activity within the brainstem is quite complex and beyond the realm of this discussion. Suffice it to say that fibers travel via the vestibulospinal tract (regulation of muscle tone), vestibulocerebellar tract (coordination), reticular formation (regulation of consciousness), and medial longitudinal fasciculus (eye movement).9–11
When the vestibular apparatus is rendered dysfunctional such as by inflammation or neoplasm, a patient may complain of vertigo, a symptom specific to disease of the vestibular system. Vertigo is described as a hallucination of movement resulting in a sensation of turning, spinning, falling, or rocking. Patients may complain that the “world is spinning around me.” This is often associated with other visceral disturbances, such as nausea, vomiting, sweating, and tachycardia. Vertigo is to be distinguished from dizziness, a sensation of light-headedness, giddiness, faintness, or unsteadiness, which is a nonspecific complaint and can be due to a variety of systemic disorders. Peripheral vertigo refers to disease of the labyrinth, vestibular nerve, or cerebellopontine angle. Central vertigo implies disease of the brainstem, including the vestibular nuclei and vestibular pathways.16
The Cochlea
Hearing arises from the capability to transform mechanical energy to electrical energy.17 The external ear collects and directs sound waves to the tympanic membrane. The middle ear converts mechanical motion arising from the tympanic membrane pulsations and transmits it to the fluid in the vestibule via the lever effect of the ossicular chain. The inner ear, specifically the cochlea, transforms fluid motion into electrical energy.
The cochlea is a coiled structure consisting of 2 1/2 to 2 3/4 turns (Fig. 5.1C, Fig. 5.1D, Fig. 5.3A, Fig. 5.4A, Fig. 5.4B). If it were elongated, the cochlea would be 30 to 32 mm in length. The modiolus (Latin for “hub of a wheel”), the central bony axis of the cochlea, is composed of spongy bone. A central bony defect within the core of the modiolus contains the cochlear nerve (Fig. 5.2D, Fig. 5.2E, and Fig. 5.2F).The modiolus is normally a robust three-dimensional (3D) structure and should never appear flattened or asymmetric at CT or magnetic resonance imaging (MRI). The normal modiolar area is < 4 mm2. On axial MRIs, the modiolus appears as a trapezoidal, bowtie, or crown-shaped low-signal core at the cochlear apex, with thin fingerlike projections, which contains the spiral ganglion (cell bodies of the cochlear nerve).18,19 Projecting outward from the modiolus throughout its length, similar to the head of a screw, is a thin bony plate referred to as the osseous spiral lamina (OSL) (Fig. 5.2E). The fibers of the vestibulocochlear nerve fan out in spiral fashion from the modiolus to pass into a channel near the root of the osseous spiral lamina, the canal of Rosenthal (Fig. 5.2F). The OSL encircles the modiolus and projects outward to the outer cochlear wall, where it attaches to the spiral ligament. The OSL encloses the cochlear duct (scala media), which contains the organ of Corti, and separates the scala vestibuli from the scala tympani.3 The interscalar septum (ISS) also projects outward from the modiolus. The ISS is distinguished from the OSL as it is much thicker (more easily appreciated at CT and MRI), does not contain neurovascular elements, and separates the apical from the middle (and middle from basilar) turn of the cochlea. Defects in the ISS between the middle and apical turns are not uncommon (scala communis) (Fig. 5.2G).3
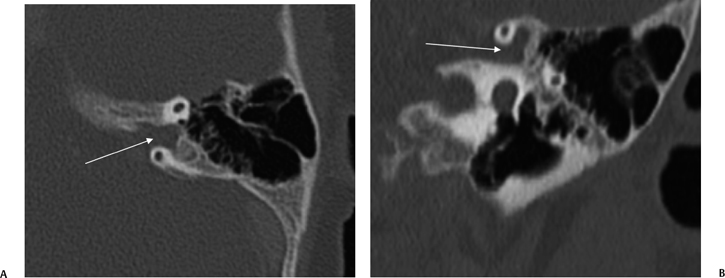
Fig. 5.6 Large petromastoid canal, child. (A) Axial and (B) coronal computed tomography images reveal that the petromastoid canal is very large (arrows). This is a normal variant in infancy and early childhood.
The fluid-filled spaces of the cochlea (endolymphatic and perilymphatic labyrinth) are composed of three parallel spiral chambers contained within the bony shell of the cochlea: an outer anterior scala vestibuli (ascending spiral) and inner posterior scala tympani (descending spiral) surround the central cochlear duct (scala media) (Fig. 5.1D). The scala vestibuli and scala tympani contain perilymph, and the scala media contains endolymph. The cochlear duct is separated from the scala vestibuli by the vestibular (Reissner’s) membrane and from the scala tympani by the basilar membrane.6,13 The organ of Corti resides within the cochlear duct on the basilar membrane. The tectorial membrane is adherent to the roof of the organ of Corti, interposed between this structure and the endolymph. As indicated in the previous paragraph, the OSL encompasses the cochlear duct (scala media) and thereby separates the cochlea into an anterior scalar chamber (scala vestibuli and scala media) and posterior scalar chamber (scala tympani). The OSL is tethered to the outer cochlear wall by means of the spiral ligament, which is the periosteal covering of the cochlear duct. The stria vascularis is the inner lining of this ligament containing numerous capillary loops and small blood vessels.
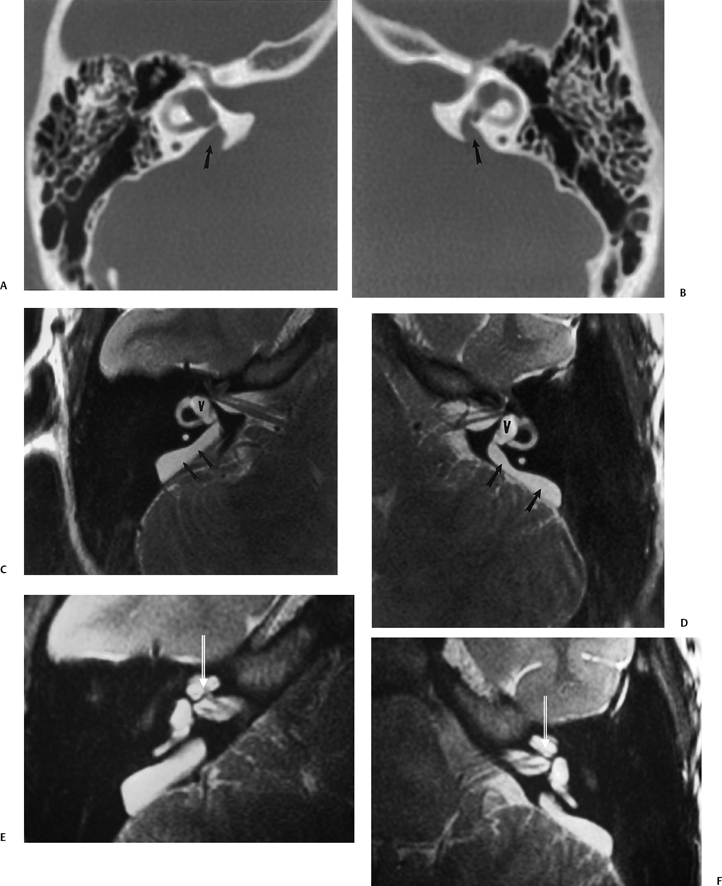
Episodic movement of the stapes results in direct transmission of fluid waves via the oval window (Fig. 5.4D) through the vestibule and subsequently to the cochlear recess, which lies on the medial wall of the vestibule (Fig. 5.1). The cochlear recess communicates directly with the scala vestibuli. Hence, the sound waves are transmitted directly to the perilymph of the cochlea as fluid waves. These fluid waves are transmitted through the Reissner’s membrane into the endolymph of the cochlear duct. This causes displacement of the basilar membrane, which stimulates the hair cell receptors of the organ of Corti. The movement of these hair cells generates the electronic potentials that are eventually converted into action potentials in the cochlear nerve fibers. It is interesting that the entire fluid volume of the perilymphatic spaces of the inner ear is only 0.2 cc, yet without it hearing would not be possible.1 The perilymphatic waves are transmitted to the apex of the cochlea (helicotrema) via the scala vestibuli to the scala tympani and eventually dissipated at the round window, which has a flexible diaphragm. The round window is located 2 mm posterior and 2 mm inferior to the oval window (Fig. 5.3C, Fig. 5.4C, Fig. 5.7A, and Fig. 5.7B).
The basilar membrane varies in width and tension from base to apex. Hence, different portions of the membrane respond to different auditory frequencies: higher frequencies closer to the base, lower frequencies closer to the apex. Sensorineural hearing loss may be categorized as sensory (cochlear) loss and neural (retrocochlear) loss. Retrocochlear hearing loss implies involvement of either the cochlear nerve or the cochlear nuclei. Defective function of the cochlea results in sensory (cochlear) loss.11
Vestibular Aqueduct and Endolymphatic Duct System
Vestibular Aqueduct
The vestibular aqueduct (VA) is a bony channel originating from the posterosuperior portion of the vestibule near the common crus. It courses posteriorly, laterally, and inferiorly along the posterior petrous surface.3 The vestibular aqueduct widens as it approaches its external aperture at the posterior margin of the petrous apex.20 In the first 20 weeks of fetal life, the course of the VA is straight and parallel to the common crus.
As the posterior fossa begins accelerated growth in the final 20 weeks of gestation, the VA and the contents assume its adult J-shaped configuration.3 The length of the VA is determined by the degree of adjacent pneumatization but is generally 6 to 12 mm (Fig. 5.4C and Fig. 5.5C).21 The vestibular aqueduct is considered large when it measures > 1.5 mm in width at the midpoint between the common crus and the external aperture.22–24 Due to its oblique nature, only segments of the normal VA are consistently seen with axial CT imaging; however, this is usually sufficient to gain an impression of the overall caliber (Fig. 5.2F). Multiplanar reformations using volume acquisition CT particularly in the sagittal or parasagittal plane may be more helpful in ascertaining the complete course of the VA.
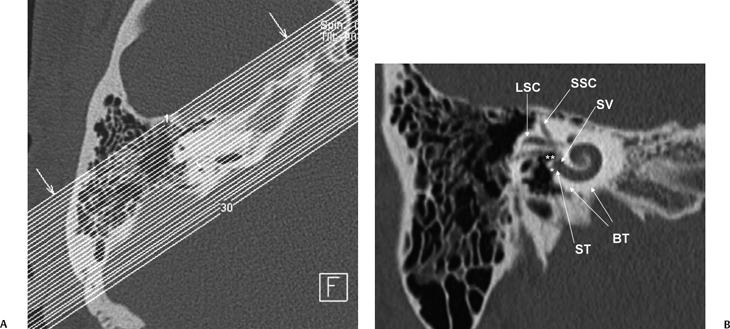
Fig. 5.7 Stenver computed tomography image. (A) Plane of section for Stenver’s projection. (B) Stenver’s view reveals oval window niche (**) leading to the scala vestibule (SV) of the basilar turn of the cochlea. Round window niche (*) leads to the scala tympani (ST) of basilar turn of the cochlea (BT). This image plane is useful in evaluating the postoperative appearance of the cochlear implant, which is inserted into the scala tympani via either the round window or the adjacent cochleostomy. LSC, lateral semicircular canal; SSC, superior semicircular canal.
Endolymphatic Duct System
The VA houses the ELD. The term endolymphatic duct system (EDS) has been advanced. The EDS is divided into an endolymphatic duct (ELD) and an endolymphatic sac (ELS) (Fig. 5.1B).
The ELD arises from the union of the utricular and saccular ducts and, after a right-angle turn, courses within the VA and emerges from an aperture along the posterior surface of the petrous pyramid.14,25 Identical to other portions of the membranous labyrinth, the ELD contains endolymph and is bathed in perilymph. The ELD is also surrounded by loose connective tissue, which is continuous with the adjacent periosteum. The ELD can be further divided into two distinct sections, the sinus and the isthmus. The sinus (horizontal) portion of the ELD (also referred to as the endolymphatic sinus) is proximal to the origin of the VA and communicates directly with the saccular and utricular ducts. The isthmic (vertical) portion is the continuation of the sinus portion and represents the intraosseous segment of the ELD housed by the VA. Histo-logically, the lining epithelium of the ELD is similar to the utricular and saccular ducts and is composed of simple squamous or low cuboidal cells.
The ELD terminates as the ELS, which is relatively large (10 to 15 mm) and often extends inferiorly along the posterior petrous surface for a significant distance.14 The ELS has a larger intradural portion and a smaller intraosseous portion. The ELS is also described as having proximal (rugose) and distal (smooth) segments. The rugose portion of the ELS is continuous with the distal ELD and consists of a complex network of interdigitating canaliculi and crypts lined by highly differentiated epithelium consisting of irregularly dispersed tall, cylindrical cells. This segment of the ELS lies within a dural sleeve in the foveate impression, which is situated along the posterior aspect of the petrous bone, partially covered by a scale of bone, the operculum.3 The distal (smooth) segment of the ELS is lined by cuboidal epithelium and situated between the periosteal portion of the dura and the dura proper. The rugose portion is important for normal endolymph resorption and for the digestion of foreign bodies. The smooth portion is thought by many to be involved in pressure equalization between the cerebrospinal fluid (CSF) and the endolymph, similar in function to the contents of the CA. The ELD/ELS quite possibly plays a role in autoregulation of the inner ear ion and fluid balance.26,27
The presence of cellular debris, macrophages, and a variety of blood cells (predominantly leukocytes) in the lumen of the EDS suggests an active role in the immune system of the inner ear as well.
The arterial supply of the ELS and ELD originates from a dural branch of the stylomastoid artery, which, in turn, may arise from either the occipital or posterior auricular arteries.25 The supplying vessels are divided into a superficial component, which directly enter the canaliculi of the vestibular aqueduct and supplies the ELS and ELD, and a deep component, which supplies the periductal connective tissue.
Cochlear Aqueduct and Contents
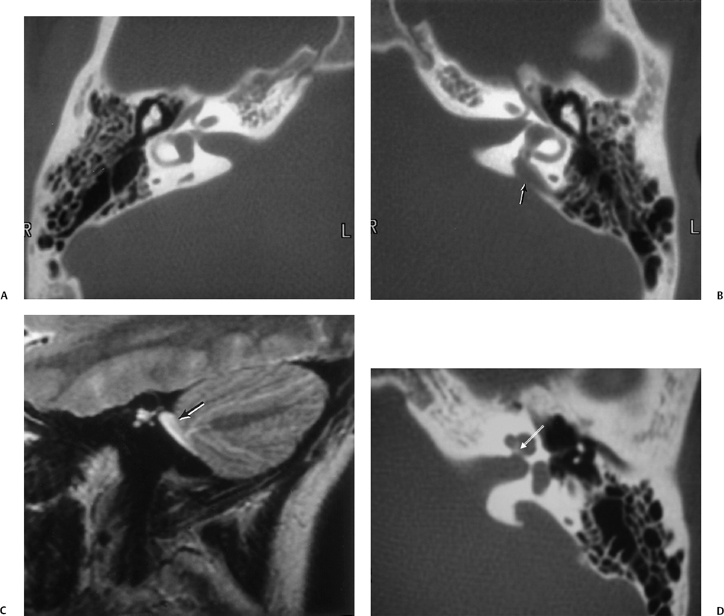
There is much confusion in the literature regarding the anatomy, function, and dysfunction of the cochlear aqueduct (CA) (Fig. 5.1).28 The CA is the narrow bony canal containing perilymph and loose connective tissue, which connects the scala tympani with the subarachnoid space. There is no membranous perilymphatic “duct” per se. The CA runs a downward oblique course and is at least partially visualized in 90% of subjects upon evaluation of axial and coronal CT images roughly parallel and immediately inferior to the internal auditory canal.29 The round window is an excellent landmark in this regard. Enhancement with gadolinium in this region often occurs but is likely of no significance clinically.30
The CA has been divided into four segments.3,13,28 From lateral to medial the CA consists of the lateral orifice, labyrinthine segment, otic capsule segment, and medial orifice (Fig. 5.2H and Fig. 5.2I). The lateral orifice is the opening into the basal turn of the cochlea, located along the anteroinferior edge of the scala tympani immediately anterior to the crest of the attachment of the round window. The otic capsule segment courses through the labyrinthine bone and never exceeds 2 mm at maximal diameter. The petrous apex segment courses through extralabyrinthine bone, which may or may not be pneumatized, and is more variable in size (average diameter of 4.5 mm). This segment widens as it approaches the funnel-shaped medial orifice, which opens into the subarachnoid space adjacent to the jugular foramen. The medial orifice of the CA is in close proximity to and should not be confused with the glossopharyngeal meatus, another funnel-shaped orifice transmitting the glossopharyngeal nerve into the anteromedial jugular foramen (pars nervosa).31
There are at least two additional closely related channels. An accessory CA transmits the inferior cochlear vein and is referred to as Cotugno’s canal. This vein terminates within either the inferior petrosal sinus or the jugular bulb.3 The tympanomeningeal fissure (Hyrtl’s fissure), also known as the second accessory canal, develops as a patent communication between the round window and posterior fossa, normally closing at 26 weeks gestation.32
This structure contains perilymph and, when patent, is much smaller than the CA. As we will see below, an anomalously patent Hyrtl’s fissure is a potential site of congenital CSF fistulization.
The function and potential dysfunction of the CA have long been a subject of debate. The CA certainly provides at least a potential conduit between the posterior fossa and inner ear. The narrow diameter of the CA is thought to buffer the inner ear from the wide pressure variations present within the posterior fossa subarachnoid spaces.28
A patent CA has been shown to permit transmission of bacterial infection between the subarachnoid space and the inner ear, which is a cause of labyrinthitis superimposed on meningitis. In theory, meningitis could be a complication of labyrinthitis via the same pathway. Hearing loss has been noted in several patients following subarachnoid hemorrhage and aneurysm surgery. It has been postulated that loss of CSF during operation results in diminished CSF pressure, which is transmitted to the perilymph via the CA, resulting in the release of perilymph into the subarachnoid space. Hemolabyrinth caused by an inflow of subarachnoid blood through the CA could be an additional factor in the development of hearing loss.33 Massive perilymphatic leak as a complication of stapedectomy (stapes gusher) has historically been linked to increased inner ear pressures caused by an enlarged and overly patent CA. This has been discounted in recent years.28
Pathology
Congenital Disorders
Overview
Congenital hearing deficits may be genetic or nongenetic. Genetic causes may occur alone or in association with a systemic syndrome and are the culprit in perhaps one half of the cases of profound childhood deafness.34–36 Genetic disorders may be autosomal dominant (one parent carries the trait), autosomal recessive (both parents carry the trait), or X-linked recessive (mother carries the trait).37
Each may have variable expressivity. In the X-linked variety of transmission, only male offspring are affected. Several autosomal recessive syndromes cause membranous deformities currently beyond imaging resolution. Some of these have associated retinal manifestations as well as sensorineural hearing loss (SNHL).37–39 These include Usher’s syndrome (retinitis pigmentosa, mental retardation), Refsum’s syndrome (retinitis pigmentosa, increased serum phytanic acid), and Cockayne’s syndrome (retinal degeneration, dwarfism). X-linked hypophosphatemic osteomalacia is also associated with SNHL.40 In these patients, there are audiometric findings suggesting endolymphatic hydrops similar to Meniere’s disease.
As described earlier in this chapter and in Chapter 3, the inner ear and middle/external ear have mutually independent embryological origins. As such, one would expect that these segments of the ear would rarely be affected together. In actuality, for reasons unknown, such anomalies do coexist on occasion. Inner ear deformities occur in perhaps 10% of patients with external auditory canal (EAC) atresia, and middle/external ear anomalies are not rare in association with inner ear dysplasias (Table 5.3).41
Nongenetic
|
Genetic
|
Bony Labyrinth
|
Membranous labyrinth only
|
Malformations Isolated to the Membranous Labyrinth
A variety of developmental malformations is limited to the membranous labyrinth and result in congenital hearing deficit. The bony labyrinth is spared because the insult occurs later in development after formation of the bony labyrinth and prior to full maturation of the sensory epithelium, particularly the organ of Corti, which occurs between weeks 26 and 28 of gestation. These dysplasias are, as of this writing, beyond the limits of current imaging methods, but several of the more common varieties are included here for completeness, specifically Bing–Siebenmann deformity, Scheibe syndrome, and Alexander syndrome. The ability to detect membranous defects may improve as technical imaging advances continue. Recent advances now permit slice thicknesses to be as thin as 0.3 mm. As the technology develops, it may be possible to visualize the components of the cochlear duct and identify structures such as Reissner’s membrane and the basilar membrane.
Bing–Siebenmann deformity is complete membranous labyrinthine dysplasia. This entity is extremely rare and may occur either as an isolated event or associated with the Jervell and Lange–Nielsen syndromes (concomitant cardiac dysfunction).42
As discussed earlier, the utricle, SCC, and ELD originate from the embryologically older pars superior, and the cochlea and saccule originate from the pars inferior. Scheibe syndrome (membranous cochleosaccular dysplasia) results from maldevelopment of the pars inferior and is believed to be the most frequent cause of congenital deafness.42–44 Pathologically, the organ of Corti is either partially or completely missing. The cochlear changes tend to be more severe in the basilar turn and gradually lessen toward the apical turn. The cochlear duct is typically collapsed, and Reissner’s membrane is deformed. The saccule is collapsed and contains degenerated sensory epithelium. The utricle is normal. The hearing loss may be unilateral or bilateral.
Alexander syndrome is a membranous dysplasia localized to the basilar turn of the cochlea. This results in familial high-frequency SNHL.45–47
Malformations of the Membranous Labyrinth and the Bony Labyrinth
Congenital malformations that affect the development of the otic capsule result in malformations of both the membranous and bony labyrinth. These malformations can be detected with current imaging methods, and the radiologist should be familiar with these disorders. The majority of congenital malformations asymmetrically affect both ears. In cases where only one ear has a radiologic abnormality, there is an 50% likelihood that the uninvolved ear will have normal hearing. The clinical findings are highly variable. Some individuals maintain normal or slightly diminished hearing, some patients experience progressive deafness, and some are deaf at birth. About 20% of patients will have vestibular symptoms.
There is a wide variety of inner ear malformations. Because the spherical otic vesicle forms three “buds,” which give rise to the cochlea, vestibule, and semicircular canals/endolymphatic apparatus, congenital inner ear malformations may affect one or more of these structures.
In previous editions, we have emphasized the overuse of the term Mondini malformation, which has in the past been used in the literature to describe virtually any malformation short of complete aplasia. Since the last edition, there have been several additional advances in classification. Perhaps most important is the use of the term incomplete partition (IP), which refers to the configuration of the cochlear turns and loss of internal architecture.
In this section, inner ear deformities are classified in order of descending severity (and later embryological insult): Michel deformity, cochleovestibular aplasia, common cavity, IP-1 (cystic cochleovestibular malformation), cochleovestibular hypoplasia, and IP-2 (Mondini).
Developmental anteromedial migration of the facial nerve canal is consistently found when the rostral growth of the cochlea is either absent or stunted.48 This occurs in most of the anomalies described in this section. The notable exception is IP-2, the classic Mondini deformity. In this circumstance, the cochlear volume is sufficient to preclude this migration (Table 5.4 and Fig. 5.8).

Michel Aplasia
This entity is likely due to a developmental arrest of otic placode development at the third gestational week before the formation of the otic vesicle, resulting in complete labyrinthine aplasia.42,49–51 This may manifest as complete absence of the inner ear or a nondescript dense bony remnant. In either case, the middle and external ear is typically entirely intact. It is extremely rare, constituting less than 1% of all congenital inner ear malformations.
Michel’s aplasia has been associated with anencephaly, thalidomide exposure, and external ear malformations. The diagnosis is relatively easy when inner ear structures are entirely absent.52 When dense bone is present, differentiation from diffuse labyrinthine ossification (most commonly due to childhood meningitis) may be a problem. In Michel’s deformity, the lateral wall of the inner ear remnant is flat in contradistinction to labyrinthine ossification, in which there is preservation of the normal convexities, particularly that subtended by the lateral semicircular canal (Fig. 5.9 and Fig. 5.10).53,54
Cochlear Aplasia
This is a very rare deformity (< 3% of cochlear malformations) characterized by complete absence of the cochlea (Fig. 5.11). The vestibule and semicircular canals are somewhat developed but typically deformed. This malformation can be explained by an arrest in the development of the otic placode during the end of the third week, but continued, albeit abnormal, development of the buds of the vestibule and semicircular canal.55 This malformation may also be confused with labyrinthitis ossificans. Ears with cochlear aplasia are devoid of auditory function.
Common Cavity Deformity
This malformation is characterized by confluence of the cochlea, vestibule, and semicircular canals, resulting in a single common cavity. This entity is relatively common, constituting 26% of cochlear malformations. The malformation can be explained by an arrest in the development of the otic vesicle during the fourth week of gestation after differentiation of the auditory (otic) placode into the otocyst, but before differentiation of the otocyst into the primordia of the cochlea, vestibule, and semicircular ducts.42,55 The result is an ovoid cystic inner ear lesion without internal architecture (Fig. 5.12). The size of this cyst varies significantly. Smaller cavities probably represent earlier developmental arrests. Cavity size averages 7 mm vertically and 10 mm horizontally.51 The internal auditory canal may be large or small depending upon the size of the cyst (Fig. 5.13, Fig. 5.14, and Fig. 5.15). There is a risk of gusher if the middle ear/oval window is surgically explored because the lateral aspect of the IAC is defective, resulting in increased perilymphatic pressure. Histological examination has revealed some differentiation of the organ of Corti scattered along the cyst wall. However, the overall neural population is usually sparse or absent. Sagittal high-resolution T2-weighted MRIs (T2WIs) in true common cavity malformations have demonstrated an absence of the cochlear nerve, precluding cochlear implantation.
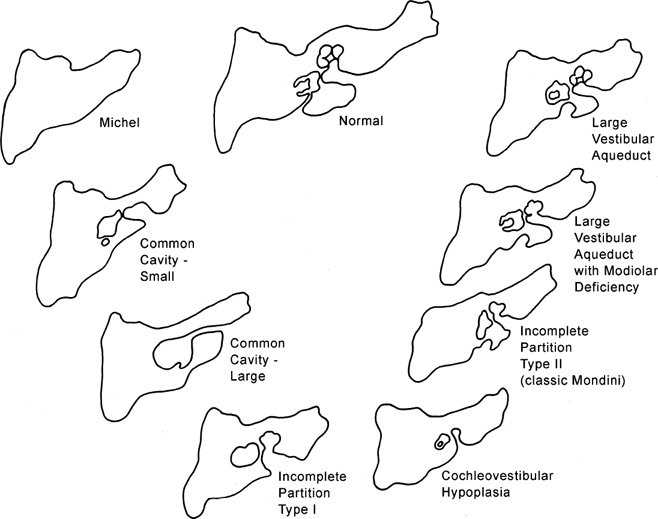
Incomplete Partition Type 1
IP-1 (cystic cochleovestibular malformation) consists of a completely unpartitioned, empty, and cystic-appearing cochlea most likely resulting from an arrest in otic placode development in the fifth week of gestation.51,56 The vestibule is always grossly dilated but distinguishable from the cochlea and as such is one step more organized and differentiated than the common cavity deformity described above. The cochlea and vestibule have been described as having a “snowman” or “figure 8” shape (Fig. 5.16).57
Importantly, the overall dimensions of the cochlea and vestibule are normal in IP-1. This has been confirmed with numerous measurements. The diagnosis is based on the absence of normal internal architecture. Specifically, the modiolus is absent, which gives the cochlea a cystic appearance. The vestibular aqueduct is normal in these cases, another important differentiating point. The facial nerve is normally positioned as the inner ear volume is preserved.48 The cribriform area between the cochlea and the internal auditory canal is often defective, and all patients have a large IAC (Fig. 5.17, Fig. 5.18, and Fig. 5.19). This predisposes to an increased risk of meningitis and to perilymphatic gusher in the event of middle ear exploration.
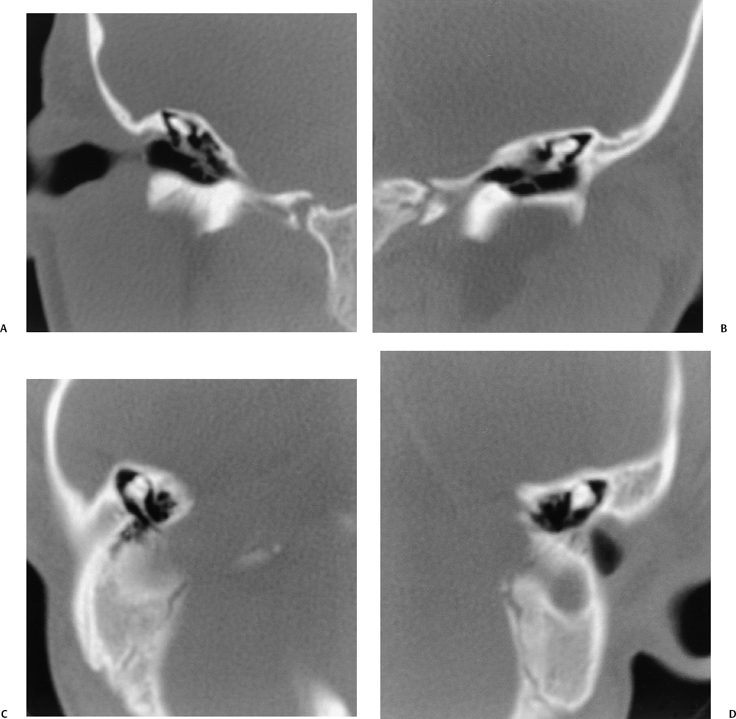
Fig. 5.9 Total labyrinthine aplasia (Michel). (A) Magnified coronal computed tomography (CT) image, right ear. (B) Magnified coronal CT image, left ear. (C) Magnified axial CT image, right ear. (D) Magnified axial CT image, left ear. Note that the middle ear and ossicular chains are developed, but that the entire labyrinth is absent. (Courtesy of Robert A. Kaufman, MD.)
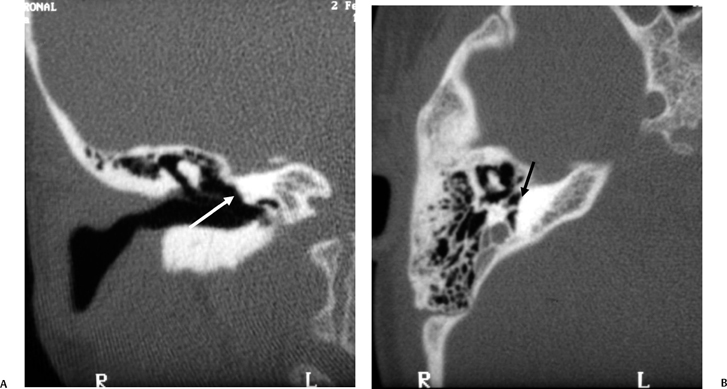
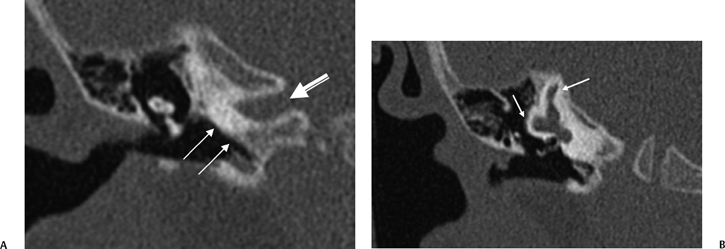
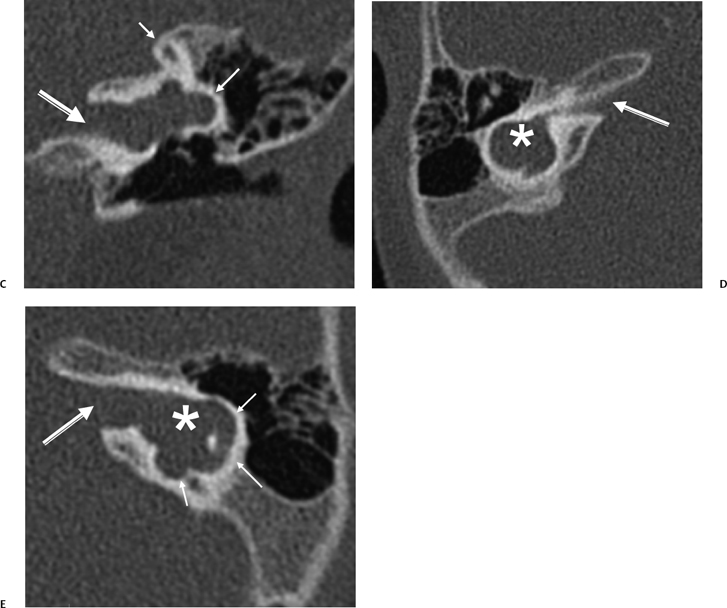
Cochleovestibular Hypoplasia
Cochleovestibular hypoplasia (CVH) represents 15% of all cochlear anomalies42,56 and is more differentiated than IP-1, but less so than IP-2. CVH is characterized by reduced cochlear length, often a single turn or less, and may be explained by a developmental arrest occurring during the sixth week of gestation. The cochlea and vestibule are differentiated from each other (Fig. 5.20). The accompanying vestibule is hypoplastic or possibly even absent. The IAC dimension is usually normal or perhaps slightly smaller than normal (Fig. 5.21). The vestibular aqueduct is normal. The internal architecture of the cochlea is variable. Hearing deficit is also variable and reflects the degree of membranous labyrinthine development. Some observers have advocated measurement of cochlear height on a routine basis, as relatively subtle cochlear hypoplasia is sometimes overlooked on initial inspection.22 A cochlear height of less than 4.35 mm is beyond two standard deviations of normal and has a high predictive value for sensorineural deficit. High-resolution T2WIs reveal variable appearance of the cochlear nerve, which may be normal or hypoplastic. As indicated elsewhere in this volume, a thorough evaluation of the cochlear nerve should be performed in patients who are potential CI candidates.
Incomplete Partition-2
IP-2 (classic Mondini malformation) can be attributed to an arrest in inner ear development during the seventh week of gestation.42,55,56 The internal architecture is more developed than IP-1. Specifically, the defect is limited to the apical and middle turns of the cochlea, where there is an absent modiolus and deficient interscalar septum (Fig. 5.22). This confluence results in a cystic appearance at the cochlear apex only. The overall dimensions of the cochlea and vestibule are normal, but the cochlea is limited to 1.5 turns. Importantly, the basilar turn of the cochlea is normal.58 The development of the organ of Corti and auditory neural population is variable, and as a result, the hearing loss ranges widely in these patients from normal to profound. Hearing loss is often fluctuating in these patients similar to that which occurs in isolated large vestibular aqueduct syndrome (LVAS). Successful cochlear implantation has been performed in several of these cases.
Vestibular prominence is variable and often minimal (Fig. 5.23). The semicircular canals are normal. As the development of the vestibular aqueduct begins just before the seventh week of gestation, the VA is always large in these patients, an extremely important differentiating point (Fig. 5.23, Fig. 5.24, and Fig. 5.25). Roughly, half have a large IAC.
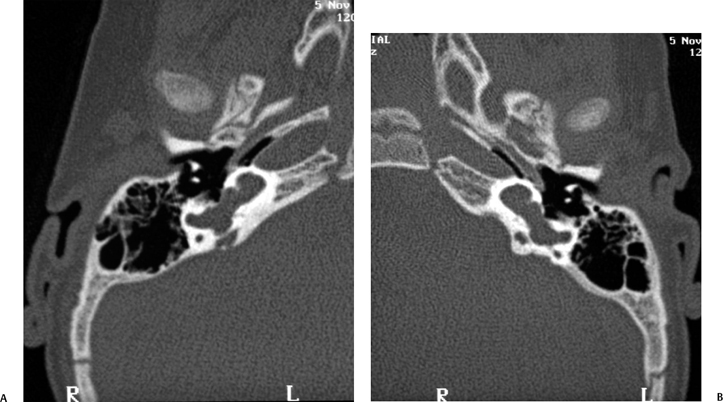
Fig. 5.12 Common cavity deformity. (A,B) The cochlea, vestibule, and semicircular canals appear as one common cavity without partition. The process is bilateral and symmetric. (Courtesy of Joel Curé, MD.)
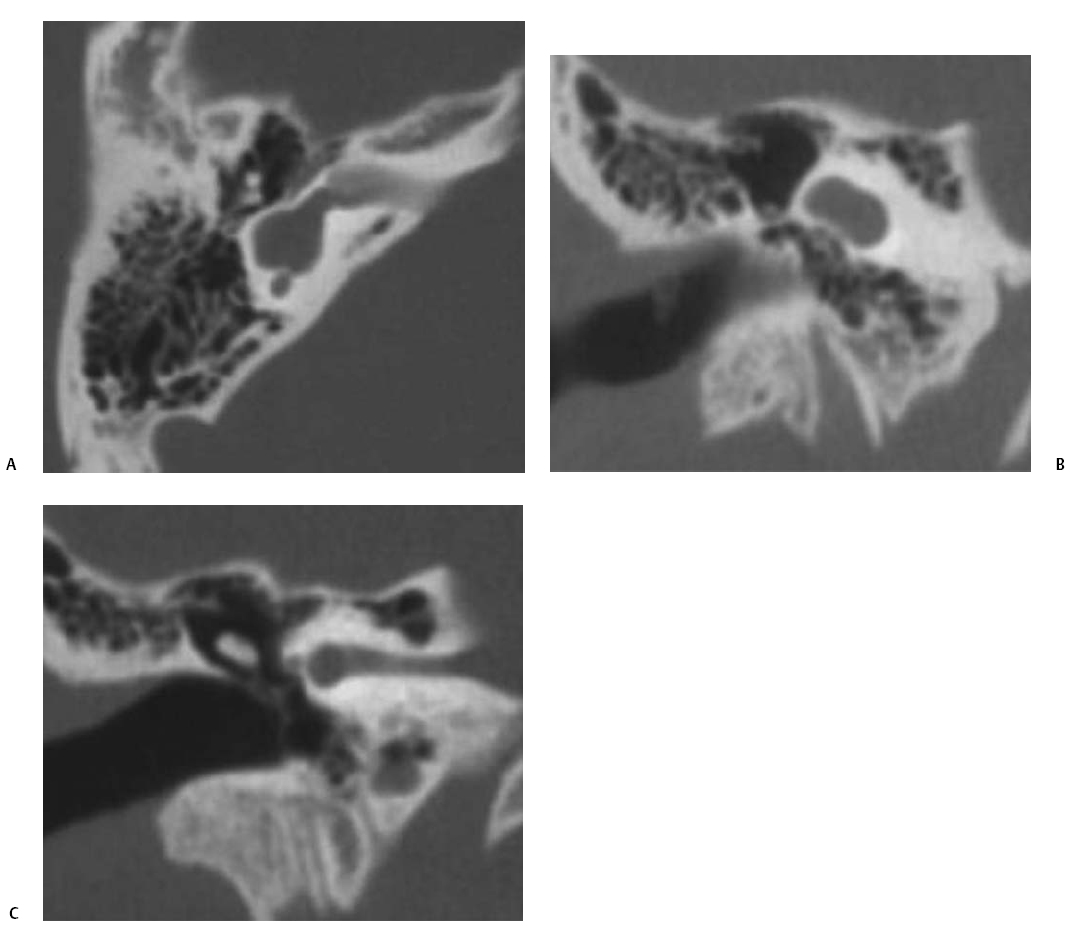
Fig. 5.13 Common cavity deformity. (A) Magnified coronal computed tomography (CT) image, right ear. (B) Magnified axial CT image, right ear. The cochlea, vestibule, and semicircular canals conform to a single cystic cavity without any evidence of partition. The middle ear and mastoid, including the ossicular chain (arrow), have developed normally.
The triad of (1) normal basilar turn/cystic cochlear apex, (2) large vestibule with normal SCCs, and (3) large VA/ELD and ELS is the precise list of conditions of a patient that were first described by Carlo Mondini in 1791 in a scientific report written in Latin, entitled “The Anatomic Section of a Boy Born Deaf.”59–61 All subsequent discussions of Mondini deformity should be limited to patients with this triad (IP-2).
As the large vestibular aqueduct is a hallmark of this disorder, there is considerable overlap between IP-2 and the large vestibular aqueduct syndrome. Quite possibly these disorders are a developmental continuum.
Large Vestibular Aqueduct Syndrome/Large Endolymphatic Duct and Sac Syndrome
Historical Significance and Pathophysiology The most common imaging finding in individuals with sensorineural deficit dating to infancy or childhood is the LVA associated with the large endolymphatic duct and sac (LED/LES). There is a high incidence of bilaterality, perhaps 90%, but findings are commonly asymmetric. This is important when cochlear implantation is contemplated, as the surgeon will want to know which ear is the most normal.62,63 As with IP-2, the developmental insult is likely in the seventh week of gestation; however, recent data suggest that the vestibular aqueduct grows throughout embryonic life and perhaps postnatally as well. This indicates that LVAS may be, to some extent, an acquired deformity rather than a stable congenital malformation.64 There is a slight female predominance. The pattern of inheritance is most likely autosomal recessive although a smaller component of autosomal dominant or multifactorial inheritance may exist (Fig. 5.26).65
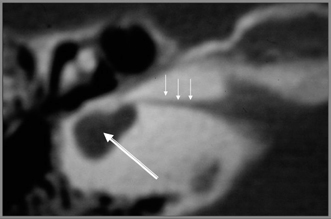
Fig. 5.14 Common cavity, small caliber. Axial computed tomography reveals that the inner ear consists of a small cystic cavity (large arrow). The internal auditory canal is extremely narrow (small arrows). See text. (Courtesy of Jan Casselman, MD.)
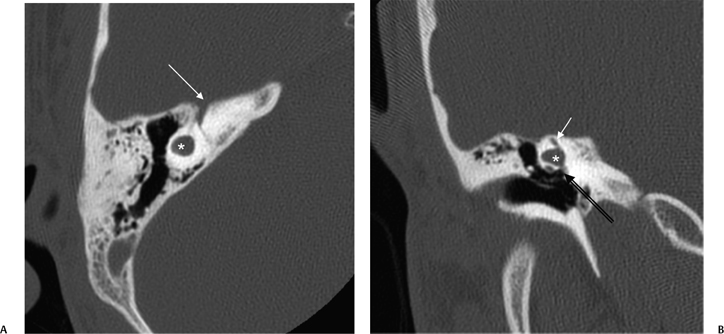
Fig. 5.15 Common cavity deformity. (A) Axial and (B) coronal computed tomography images. There is a small common cavity deformity (*) without differentiation into the cochlea or vestibule/semicircular canals, although a tiny SSC remnant (short arrow, B) is noted. Oval window (long arrow, B); note the presence of the ossicular chain. First genu of FNC (arrow, A). (Courtesy of Bernadette Koch, MD.)
The association between an LVA and SNHL was initially described by Valvassori and Clemis in 1978.24 They identified 50 patients with enlarged VAs using polytomography, coining the term large vestibular aqueduct syndrome (LVAS). Despite imaging limitations, they also astutely noted that 60% of patients with enlarged VAs had other inner ear malformations. In 1989, Levenson and colleagues identified LVAS as a distinct clinical entity.66 They determined that LVAS is a congenital anomaly of the temporal bone that predisposes patients to acquired, progressive, fluctuating, high frequency SNHL. The hearing loss may be triggered by head trauma and activity that causes sudden fluctuation in CSF pressure. Based on their own similar investigation, Jackler and De La Cruz advised patients with LVAS to avoid contact sports and activities that entail extreme barometric pressure changes.67 Harnsberger and Davidson, as well as Okamoto, noted that the bony changes of the enlarged VA are probably caused by the enlarged ELD and ELS and suggested that the name of the entity be changed to large endolymphatic duct and sac syndrome (LEDS) or, alternatively, large endolymphatic sac anomaly (LESA).18,57,68 The terminology used probably reflects which modality (CT or MRI) is used in diagnosis.
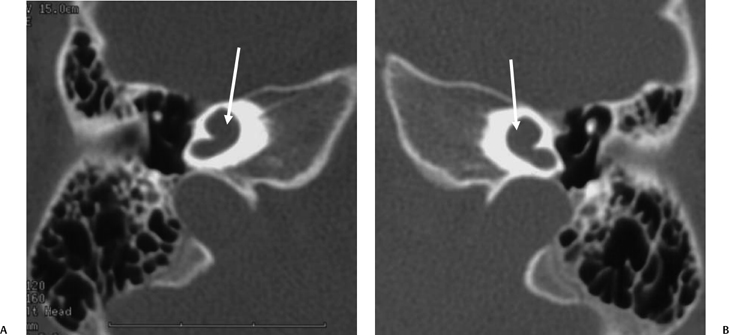
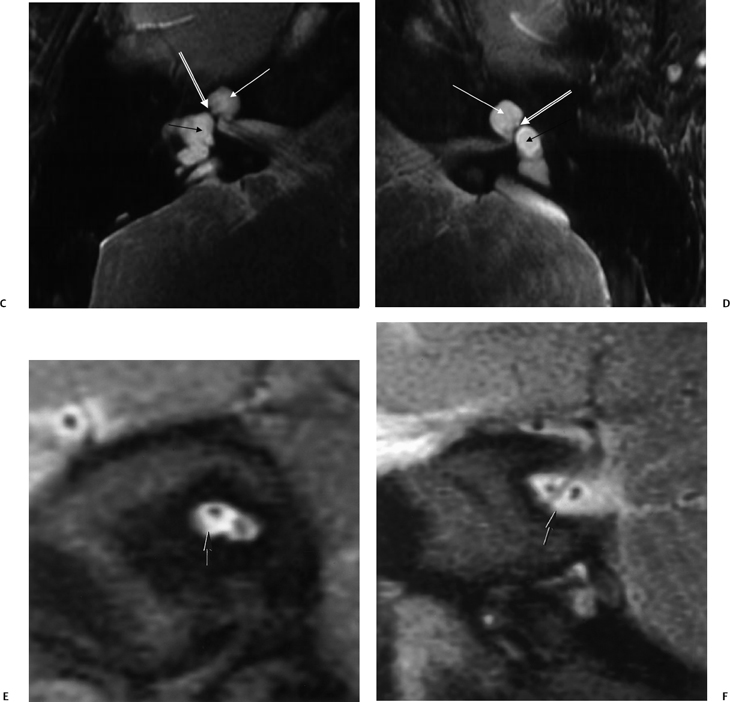
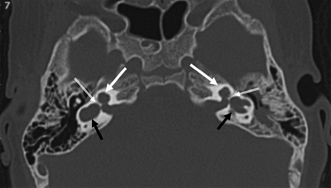
Fig. 5.17 Incomplete partition type I (IP-I) anomaly. Axial computed tomography image reveals a bilaterally symmetric dysplasia characterized by a cystic cochlea (absent modiolus) (white arrows) and cystic vestibule (black arrows). Note the well-defined partition (outlined arrows) between them, indicating that this is a step advanced from the common cavity deformity. Note that examination of the posterior petrous surface reveals no enlargement of the vestibular aqueduct, a hallmark of this deformity. (Courtesy of C. Douglas Phillips, MD.)
The precise cause of the hearing deficit in these patients is unknown; however, numerous interesting theories have been advanced. Foremost among these theories are (1) hyperosmolar proteins in the enlarged ELS reflux into the cochlear duct (scala media) through a widely patent ELD, which causes osmotic damage to the neuroepithelium; and (2) associated modiolar deficiency allows CSF pressure waves into the labyrinth, damaging hair cells in the organ of Corti.19,66–70 The latter theory was advanced more recently by the observers who recognized the high incidence of cochlear anomalies in LVAS/LEDS/LESA. Relative T2-weighted hyposignal is very commonly present in the large endolymphatic sac, likely reflecting the extremely high protein concentration known to occur in this region. This fact is more supportive of the former theory (reflux into cochlear duct).71 Internal fistulization (mixing of perilymph and endolymph) likely also plays a role in the pathogenesis of the episodes of hearing deterioration similar to that which occurs in other malformations. This explains why patients are usually instructed to avoid contact sports and activities, such as scuba diving, which result in extreme barometric pressure changes. Patients may also benefit from a no-salt-added diet.72,73 Others have suggested that a buildup of toxic metabolic by-products in the endolymph or interference with the immune response could result from a dysfunctional endolymphatic sac. Endolymphatic hydrops is an unlikely culprit, as the home-ostatic disruption would be expected to involve the apex of the cochlea and result in a low-frequency (audiometrically up-sloping) sensorineural hearing deficit. As such, endolymphatic sac shunting has fallen into disrepute; however, surgical ELS obliteration has been determined to be useful in stabilization of the progressive hearing loss associated with this disorder.67,74,75 The reader should be aware that the overall size of the VA, ELD, and ELS and the degree of modiolar deficiency (see below) do not correlate with the degree of hearing loss.76
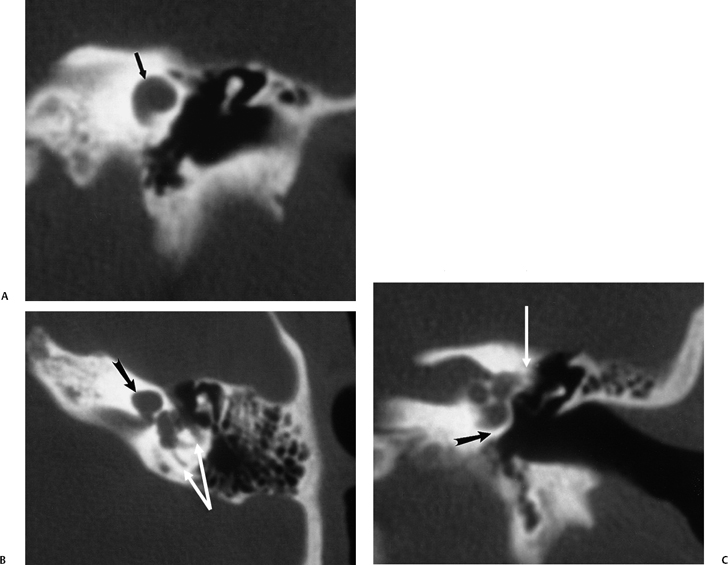
Fig. 5.18 Incomplete partition type I (IP-1) anomaly with modiolar deficiency, semicircular canal dysplasia. (A) Coronal computed tomography (CT) image at the level of the cochlear apex. (B) Axial and (C) coronal CT images at the level of the vestibule. There is a cystic cochlear apex (black arrows, A,B) resulting from modiolar deficiency, although the basilar turn of the cochlea is unremarkable (black arrow, C). There is no evidence of a large vestibular aqueduct that would indicate the classic Mondini deformity. This malformation is instead associated with hypoplasia of the semicircular canals (white arrows, B,C).
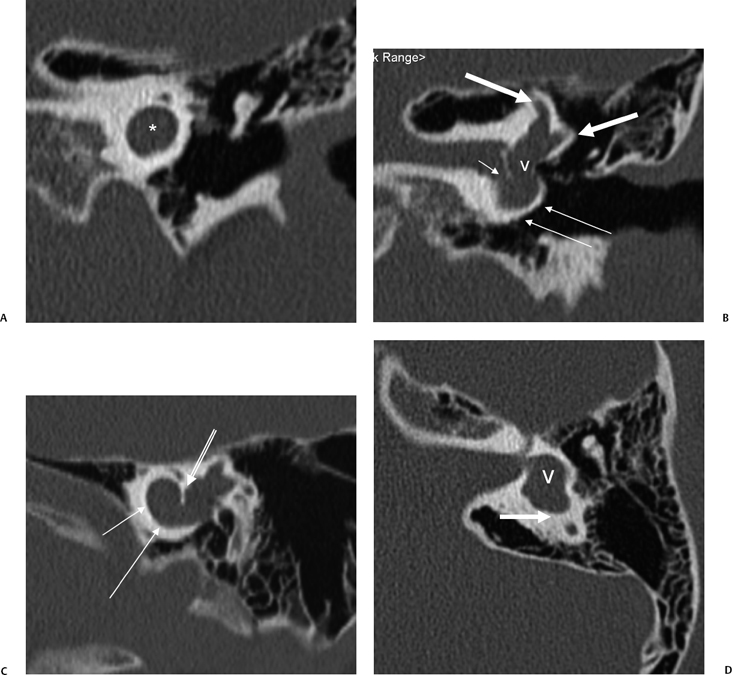
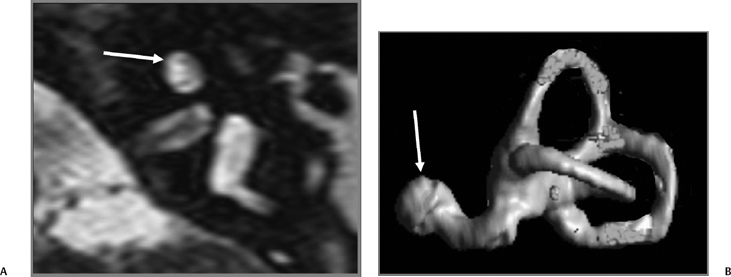
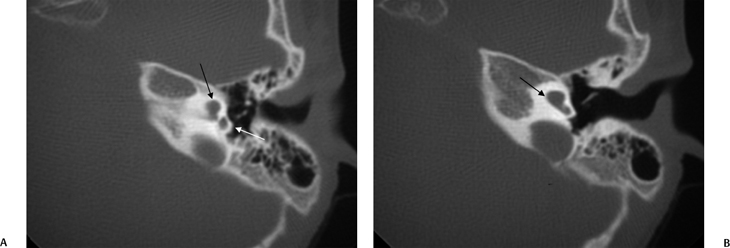
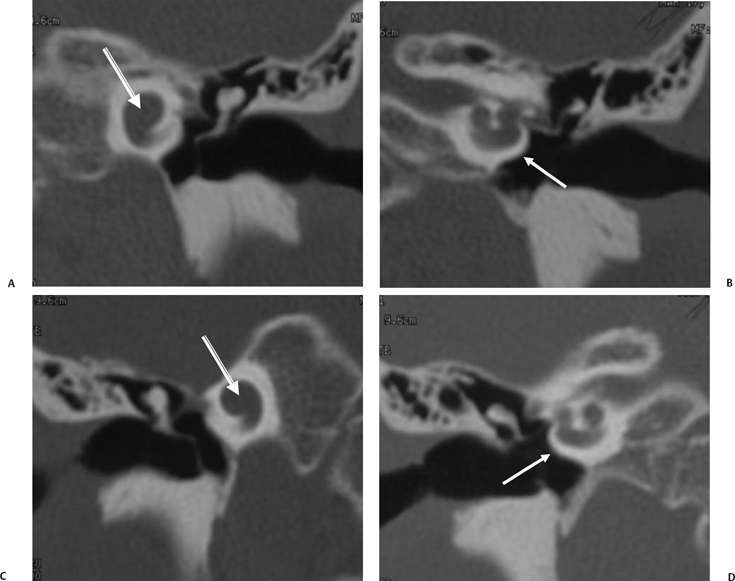
Fig. 5.22 Incomplete partition type 2 (classic Mondini deformity), bilateral. Coronal CT (A,B) left ear and (C,D) right ear reveal a cystic cochlear apex (outlined arrows); however, the basilar turn (solid arrows) is normal. The intact basilar turn is a critical differentiating point. The vestibular aqueduct was large on each side (not illustrated). Note that there is no evidence of semicircular canal development.
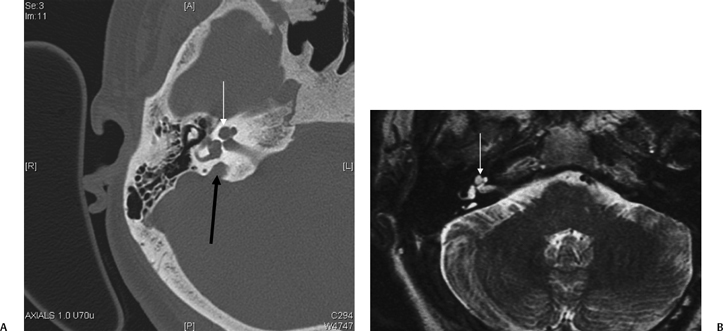
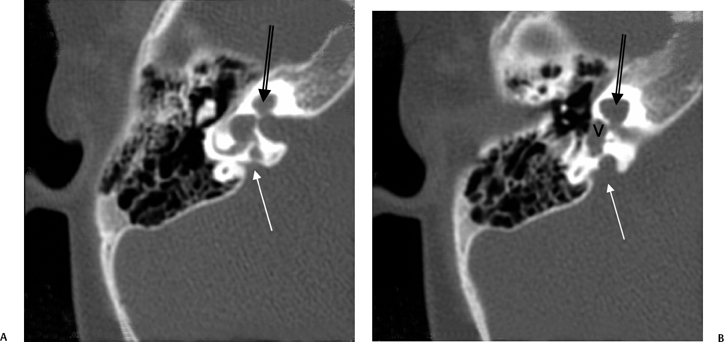
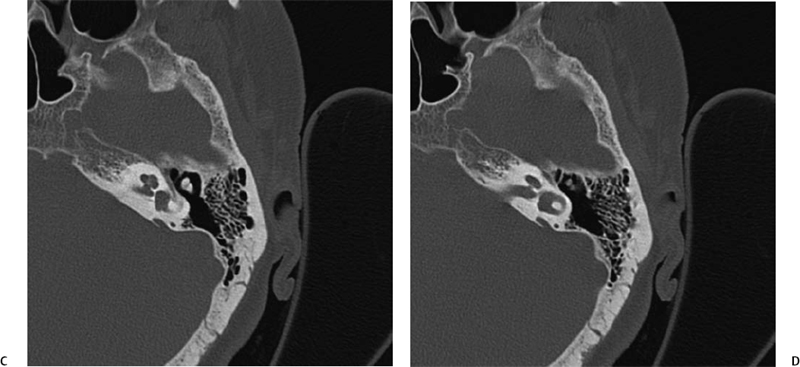
Fig. 5.24 Incomplete partition-2 (classic Mondini deformity). There is a cystic cochlear apex (black arrows), a dilated vestibule (V), and a large vestibular aqueduct (white arrows). (Courtesy of Barb Zeifer, M.D.)
Imaging Features of LVAS/LEDS The characteristic imaging features are the LVA on CT and the enlarged ELD and ELS on MRI.18,23,24,77 The VA/ELD is considered large when it measures > 1.5 mm in width at the midpoint between the common crus and the external aperture.21,23,24 The size of the ELS is highly variable and not necessarily proportional to the size of the VA. On occasion, the ELS may be massive, with impingement upon the anterolateral cerebellum, and could conceivably be confused with other pathological conditions18,78,79
There have been major advances in the evaluation of this disorder in recent years. These have revealed that vestibulocochlear abnormalities are much more common in this disorder than originally thought.18,19,80 In particular, cochlear anomalies are quite commonly appreciated with both CT and MRI, but especially with high-resolution T2WI techniques. These range from simple modiolar deficiency or scalar asymmetry to full-blown cochlear dysmorphism.18 In another recent series, all patients had, at minimum, anomalies in the modiolus of the cochlea.19 Other observers have noted that although modiolar deficiency is extremely common in this disorder, occasionally these patients do have a normal modiolus; in one series, 14% of patients had a normal modiolus.18,71,76 As indicated above in the anatomy section, the normal modiolus appears as a 3D structure on axial T2WI (and at CT) through the cochlear apex. It often has the shape of a crown, a bowtie, or a trapezoid; it should never appear flattened or decidedly asymmetric (Fig. 5.27 and Fig. 5.28).
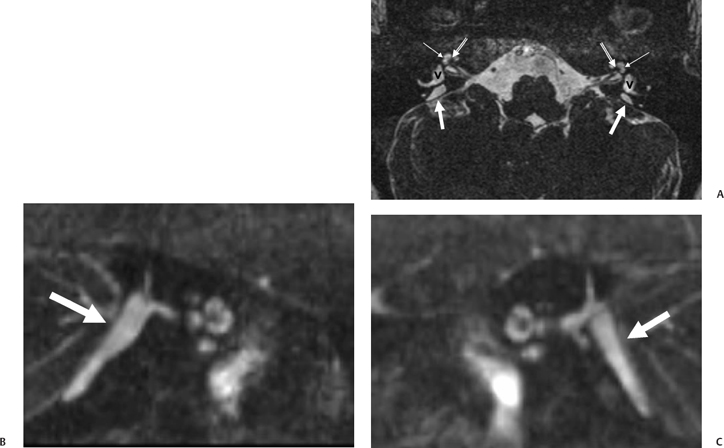
As one would expect, T1WI and T2WI relaxation times within the ELD are usually long, approaching those of CSF, reflecting fluid content.73 Reflux of hyperosmolar proteins may alter the MRI characteristics of the ELS, as high protein may shorten T1WI and T2WI relaxation times (high signal intensity on T1, low signal intensity on T2).68 Importantly, occasional patients with congenital SNHL are identified with a large ELS in the absence of an unusually large VA. Conversely, measurably large VA/ELD may be associated with a relatively unremarkable ELS.81
As noted above, the severity of the hearing loss does not correlate linearly with the size of the VA/ELD/ELS. A recent study revealed that a deformed LSCC was closely associated with the presence of vertigo in patients with LVAS.82
The LVA (and dilated ELD/ELS) may also occur in association with several of the cochlear malformations described above and is a hallmark of the classic Mondini deformity (IP-2). LVA does not occur with IP-1. It is interesting to speculate on how many patients diagnosed in the past with LVAS may actually have had classic Mondini deformity (IP-2).
As indicated elsewhere in this volume, LVAS is associated with several named syndromes. Pendred syndrome (see below) is the most notable of these. Recently, several patients with mutations in the GJB2 gene have been described with findings indistinguishable from this disorder.83
Semicircular Canal Anomalies
Overview
Our understanding of congenital malformations of the SCC has rapidly increased over the past few years. This is based on a greater knowledge of molecular genetics and advances in our ability to visualize the SCC on cross-sectional imaging. To understand the spectrum of malformations, we must first review the embryogenesis. As previously discussed, the membranous labyrinth has three subdivisions, the endolymphatic duct and sac, the pars inferior (cochlea and saccule), and the pars superior (utricle and semicircular ducts). The superior, posterior, and lateral semicircular ducts reach their adult configuration by the eighth week of gestation. Ossification of the semicircular canals occurs at approximately midgestation. The SSCC is completed first (19th week), and the LSCC is the last to completely develop (22nd week). There is consensus that because the horizontal canal is the last to develop, it is more susceptible to anomaly.
Semicircular Canal Dysplasia and Aplasia
As discussed in the above sections, the type of inner ear malformation can often be attributed to the timing of the developmental arrest. Because the cochlea and SCCs have a common progenitor, the otic vesicle, dysplastic semicircular canals should be associated with cochlear malformation. Similarly, patients with semicircular canal dysplasia should have vestibular dysplasia due to their contiguity.
However, in 1990, Parnes and Chernoff reported two cases of bilateral complete SCC aplasia associated with a normal or near-normal cochlear development.84 This is difficult to explain embryologically; however, subsequently, several other reports of isolated SSC aplasia have surfaced.5,85 This anomaly appears to be more common in males, with the average of presentation between 5 and 10 years of age. Patients often present with profound hearing loss that is difficult to explain. Affected children had some delay in supporting their head and sitting up in infancy and also had delayed milestones for walking.
These deformities are not explained by a single classification system based on embryonic arrest. Instead, altered molecular genetics is a concept gaining greater acceptance, implying that the isolated SCC aplasia may be due to specific gene mutations. Recent investigations in mice have corroborated this theory. This deformity may be an important marker identifying patients who may benefit from genetic testing.
SCC dysplasia may be sporadic or syndromic. In general, the dysplasia is more severe when syndromic. Importantly, absent semicircular canals are a hallmark of the CHARGE syndrome.86–88 Several other ear anomalies are associated with this syndrome.88 Greater than 70% will have oval/round window atresia, cochlear nerve aperture atresia, facial nerve canal anomalies, and middle ear deformity (Fig. 5.29).
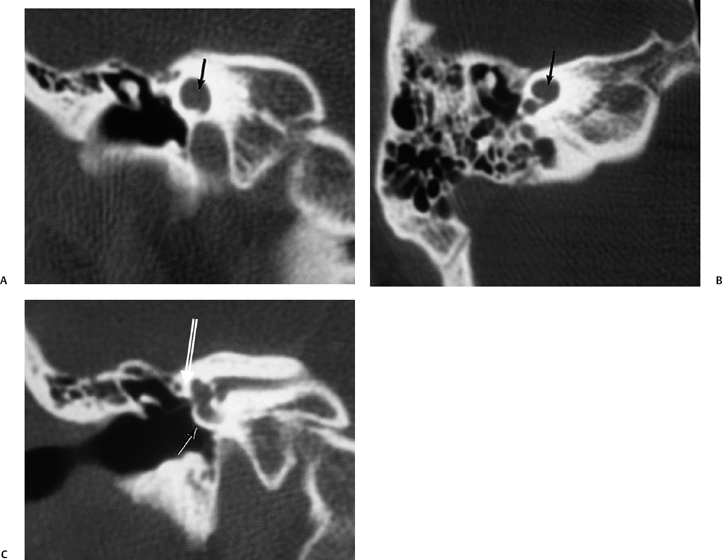
Fig. 5.29 Semicircular canal dysplasia; modiolar deficiency. (A) Coronal computed tomography (CT) image at the level of the cochlear apex. (B) Axial and (C) coronal CT at the level of the vestibule. There is a cystic cochlear apex (black arrows, A,B) resulting from modiolar deficiency, although the basilar turn of the cochlea is unremarkable (black arrow, C). There is no evidence of a large vestibular aqueduct, which would indicate the classic Mondini deformity. This malformation is instead associated with hypoplasia of the semicircular canals (white arrow, C).
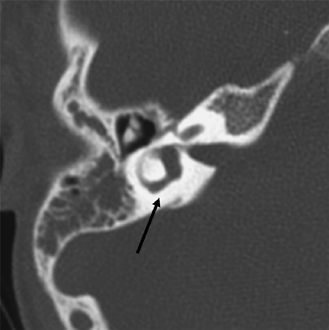
Selective aplasia of a specific SCC may also occur. Isolated lack of development of the posterior SCC has been described in Alagille’s syndrome (Fig. 5.30) (arteriohepatic dysplasia) and Waardenburg’s syndrome.89–91
Selective absence of the SSCC has been also been reported. Isolated dysplasia of the LSCC (with dilated vestibule) may be sporadic, but it also occurs in acrocephalosyndactyly type I (Apert) and Crouzon’s craniofacial dysostosis. Unilateral SCC aplasia has been diagnosed in a patient with Goldenhar’s syndrome (oculoauriculovertebral dysplasia).92 This is unusual because ear findings in this disorder typically involve structures subtended by the first and second branchial arches (middle and external ear).
Superior Semicircular Canal Dehiscence
The syndrome of superior semicircular canal dehiscence (SSCD) has been described in the literature since our last edition.93–98 This results in dizziness and vertigo, which may be triggered by straining, heavy lifting, or loud (typically low frequency) sounds. Another common complaint is autophony (reverberation when speaking, chewing, or swallowing). Hearing loss is also associated, which is typically sensorineural, but can be conductive. Men are affected more commonly than women, and presentation is often between 30 and 50 years of age. This disorder is usually unilateral; however, a case of bilateral SSCD was recently reported.99
The oval and round windows are normally the only two openings in the hydraulic system of the inner ear, and as a result, movement of inner ear fluid with stapes motion is anatomically limited although physiologically crucial to normal hearing and balance.93 The inward bulging of the oval window caused by sound-induced stapes movement is compensated by outward bulging of the round window, resulting in a precise equilibrium. Dehiscence of the bone overlying the SSSC creates a “third mobile window,” which results in aberrant movement of inner ear fluids. Loud sounds push the stapes inward, and the third window moves outward (Fig. 5.31A). This creates a wave in the perilymphatic compartment, which compresses the endolymphatic compartment, resulting in dizziness. Sound-induced vertigo, nystagmus, or both are referred to as the Tullio phenomenon. This phenomenon has been described in several other entities, including erosive cholesteatomas, otosyphilis, and in patients who have undergone artificial lateral semicircular canal fenestration, which was used in the past to treat otosclerosis prior to the development of prosthetic stapedectomy (see Chapter 3). Other types of surgery may also be causitive (Fig. 5.32). Patients may also have Hennebert’s sign, which refers to nystagmus caused by applied pressure within the external auditory canal.94 An interesting corollary in these patients is the similar symptomatology created by the Valsalva maneuver, which results in a transient increase in intracranial pressure and pushes the third window inward rather than outward. The only clinical difference is that although the plane of nystagmus remains oriented with the plane of the SSC, the nystagmus produced with the Valsalva maneuver is in the opposite direction.
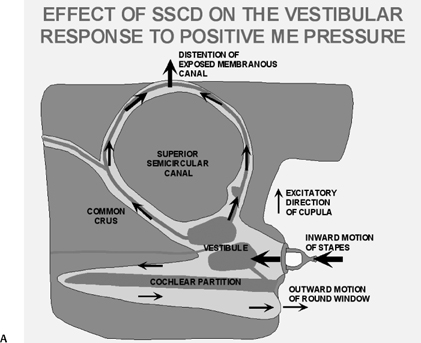
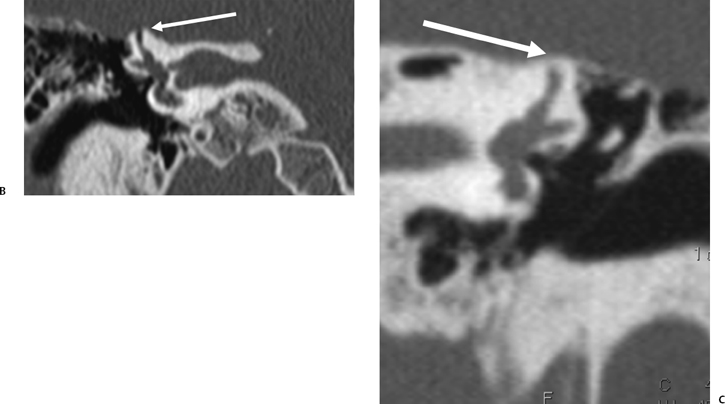
Fig. 5.31 Superior semicircular canal dehiscence syndrome. (A) Drawing illustrates the distention of the membranous labyrinth of the SSC, which occurs as a result of a loud noise. (See Color Plate Fig. 5.31.) (B) Coronal CT reveals an absent cortex of the superior semicircular canal (arrow). (C) Coronal CT, opposite ear, reveals a normal cortex (arrow). (Case courtesy of C. Douglas Phillips, MD. Drawing courtesy of H. D. Curtin, MD.)
SSCD can be identified with high-resolution CT.94–97 The imaging findings are absence of bone forming the roof of the superior surface of the canal (Fig. 5.31B,C). The roof of the SSC is composed of up to three layers: (1) otic capsule, (2) trabecular bone (may be pneumatized), and (3) cortical bone continuous with the superior surface of the remainder of the petrous pyramid (arcuate eminence). The CT diagnosis of SSCD is challenging, as even a sliver of intact bone excludes the diagnosis (“papyraceous osseous margin”). The well-known partial volume effect results when a structure is smaller and thinner than the individual voxel used to generate the CT image. As the superior surface of the canal may be extremely thin, images obtained with ≥ 1.5 mm collimation may create the illusion of a dehiscence in the normal patient. For this reason, 1.0 mm or 0.5 mm collimation is recommended. Oblique reformations further increase the positive predictive value of the examination, as the SSC plane is 45 degrees divergent from both the coronal and sagittal planes.19 As such, reformations in the planes of Stenver and Poschl may be of value in many cases (Fig. 5.5A, Fig. 5.6A, and Fig. 5.33).93,98 Both of these planes of section are 45 degrees oblique to the coronal and sagittal planes, but perpendicular to each other (Fig. 5.5). The entire arc of the superior semicircular canal is seen on one slice in the plane of Poschl. Detailed diagnosis is crucial, as treatment is craniotomy with middle cranial fossa repair by plugging or resurfacing the bony defect. Surgery is often highly successful in treating the vertigo; however, hearing may improve or worsen.100
Symptomatology identical to SSCD has been diagnosed anecdotally in patients with a number of different pathologic and imaging findings.101–103 CT performed in one patient revealed posterior SSCD caused by petrous apex cholesteatoma. Another had a subarcuate vascular malformation resulting in widening of the petromastoid canal, which caused SSCD adjacent to the common crus. As discussed in Chapter 6, the petromastoid canal is a curvilinear normal structure containing the subarcuate artery and vein, which courses between the limbs of the SCC. A third patient had classic auditory and vestibular symptoms that were proven to be caused by developmental dehiscence of the posterior semicircular canal. The pathophysiology (third mobile window) in all three of these examples was identical to classic SSCD.
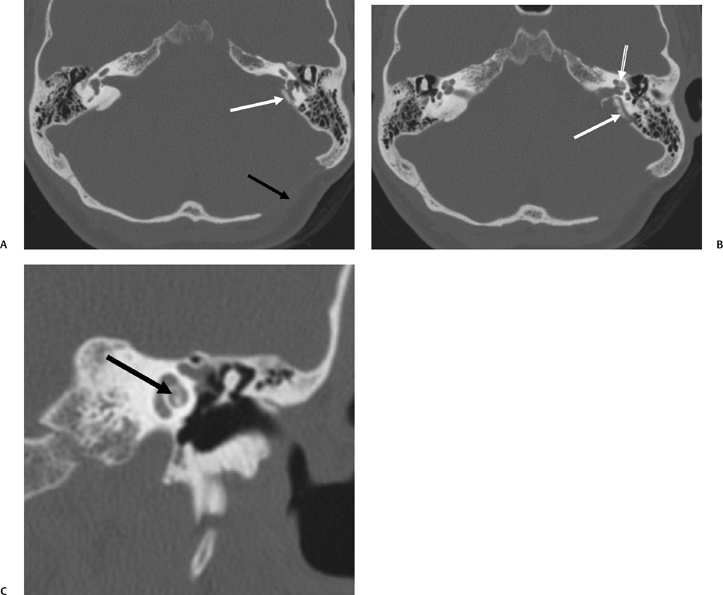
The thickness of bone between the carotid canal and the basilar cochlear turn (cochlear-carotid interval) is widely variable, and a case with completely absent bone was recently reported.104 It has been postulated that energy transmitted from artery pulsations may result in alterations of intracochlear fluid dynamics and aberrant hair cell stimulation. Tinnitus and SNHL may result. In many respects, this is analogous to SSCD as well.105 This entity is discussed further in the section on cochlear implantation.
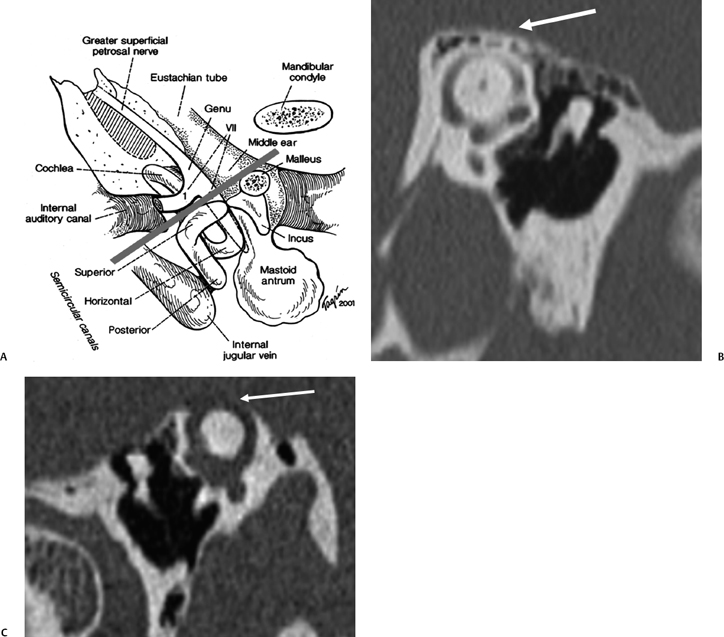
Isolated Deformity of the Lateral Semicircular Canal
As indicated in previous paragraphs, the SSCC is the first to develop, followed by the posterior and, finally, the lateral. As the LSCC is the last to develop, malformations isolated to the lateral semicircular canal are relatively common (Fig. 5.34). This deformity may be unilateral or bilateral and can be associated with sensorineural deficit, conductive deficit, or normal hearing.106 Although not associated with a positive predictive value for associated hearing loss, a normative measurement has been devised through examination of the LSCC “bony island.” This measurement is from the apex of the canal turn to the junction of the canal with the vestibule. Measurements of < 2.6 mm or > 4.8 mm are beyond two standard deviations and indicative of hypoplasia and hyperplasia, respectively.22,107
Syndromic Hearing Loss
Congenital sensorineural hearing deficit is associated with a large number of systemic eponymically designated syndromes. A complete review is beyond the scope of this text. Rather, we focus on entities for which there are established inner ear deformities detectable with current imaging methods.
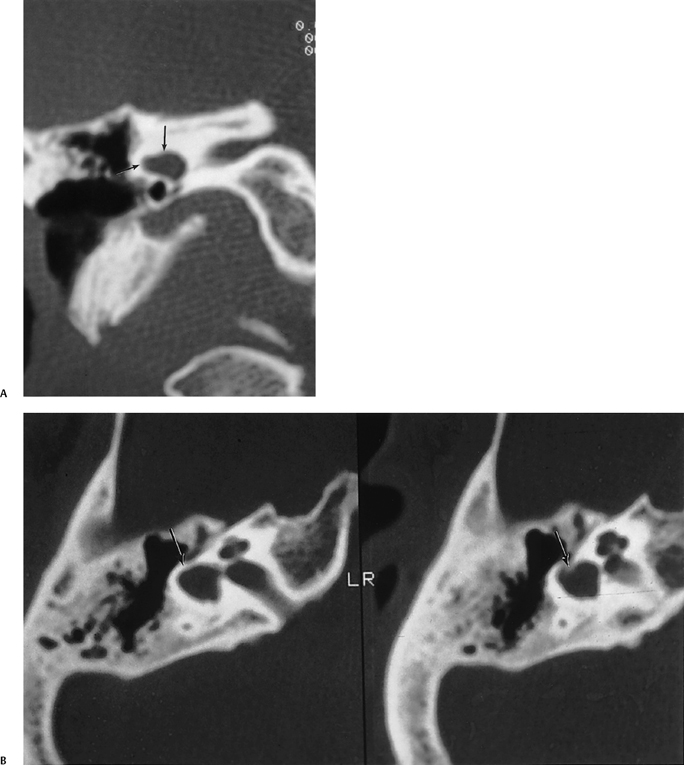
The most common syndromic cause of profound hearing loss is Pendred’s syndrome, accounting for perhaps 10% of cases. This is an autosomal recessive disorder linked to chromosome 7, which is characterized by euthyroid (although hypothyroidism may occur in some) goiter, and a positive perchlorate discharge test.108 The thyroid glands of affected individuals cannot organify iodide efficiently, and a variable amount of iodine taken up by the gland is discharged after administration of perchlorate. This constitutes the basis of the perchlorate discharge test. Hearing deficit ranges from dramatic and prelingual to slowly progressive and fluctuating and may worsen following a traumatic event or infection. The vast majority of patients have a large vestibular aqueduct (with large endolymphatic duct and sac) and a large vestibule (Fig. 5.35). Therefore, it is not surprising that patients often suffer fluctuation and sudden worsening following episodes of trauma or infection (similar to LVAS/LEDS/LESA). The apical turn of the cochlea is commonly absent. Modiolar deficiency is a constant finding, 100% in one series.108 Rarely, Pendred syndrome is diagnosed in the absence of goiter.
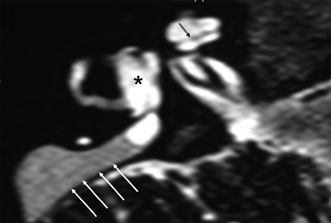
Anecdotal reports of the “Mondini type” of inner ear deformity have been reported with the syndromes of Klippel-Feil, Wildervanck, Alagille, Waardenburg, Pendred, and DiGeorge.35,44 Klippel–Feil syndrome (fused vertebrae, pectoral deformity) is associated with numerous other inner ear anomalies, including internal auditory canal duplication.109–112 Conductive hearing deficit (CHD) is more common in these patients and is secondary to anomalies of the distal incus and stapes. Wildervanck syndrome (Fig. 5.36) (cervico-oculo-acoustic) consists of the Klippel–Feil anomaly, abducens nerve dysfunction, and congenital deafness. The abducens dysfunction is believed to be supranuclear in origin and results in a distinctive eye movement disorder known as Duane retraction syndrome. The female to male inheritance ratio is 10:1. Severe anomalies including aplasia of the semicircular canals, cochlear hypoplasia, and incomplete partition type 1 have been described.50 Goldenhar’s syndrome is associated with branchial arch deformity as well as findings consistent with the Klippel–Feil anomaly. As such, they may be part of the same spectrum. Alagille syndrome (arteriohepatic dysplasia) is associated with chronic cholestasis, cardiovascular abnormality, vertebral arch defect, growth and mental retardation, and hypogonadism. Severe hypoplasia involving the posterior semicircular canal has been consistently identified. Middle ear anomalies also occur.
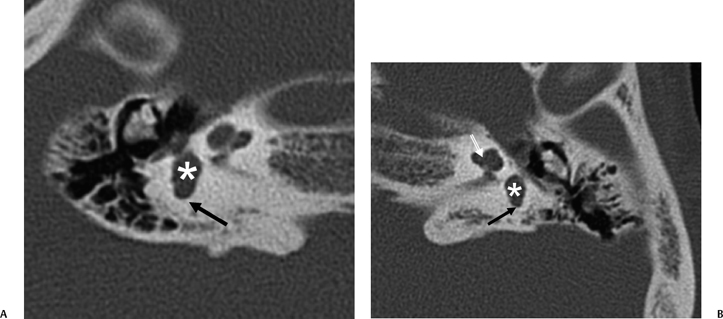
Fig. 5.36 Wildervanck syndrome (cervico-oculo-acoustic). Patient has Klippel–Feil deformity and long-standing hearing deficit. (A) Axial computed tomography (CT) image, right ear. (B) Axial CT image, left ear. The vestibule is large bilaterally (*) and there is almost complete semicircular canal (SCC) aplasia. There is minimal development of the ampullated end of the posterior SCC bilaterally (black arrows). There is modiolar deficiency appreciated on the left (white outlined arrow). Similar findings would be expected with the CHARGE association. (Courtesy of Paul Caruso, MD.)
Waardenburg syndrome (hypertelorism, iris heterochromia, white forelock) has a strong association with congenital sensorineural hearing deficit. The most common imaging finding is the large vestibular aqueduct with modiolar deficiency.113 Pathological evaluation has also revealed membranous labyrinth deformities, although a hypoplastic cochlea and posterior semicircular canal dysplasia have been reported in several of these patients as well.114
Branchio-oto-renal syndrome is well documented and features auricular deformity, hearing loss, and renal anomalies (Fig. 5.37 and Fig. 5.38). Various reports have linked cochlear hypoplasia (hypoplastic apical cochlear turn), characteristic medial facial nerve deviation flared IAC, SCC deformities, and large VA with this autosomal dominant disorder.115–118 A patulous eustachian tube is also consistently appreciated.
A large number of inner ear anomalies are described in CHARGE syndrome.87,88 SCC aplasia is virtually universal in these patients. Oval window and cochlear nerve aperture atresia is also extremely common (Fig. 5.39). Various cochlear deformities may occur, and numerous middle ear deformities are reported as well. Occasionally, IAC hypoplasia and LVAS/ELD/ELS enlargement are also seen in these patients.119
Individuals with Down syndrome (trisomy 21) are well known to have middle and external ear anomalies. Inner ear anomalies are also not uncommon and usually involve the vestibule and lateral semicircular canal, which are often hypoplastic and partially fused.120
Crouzon’s syndrome (craniofacial dysostosis) and Apert’s syndrome (acrocephalosyndactylism type I) are associated with large vestibules and short, deformed LSCC. Deformity of the middle cranial fossa secondary to the severe craniofacial asymmetry with upward angulation of the petrous apex relative to the external auditory canal is commonly associated. In point of fact, most ear symptoms in these patients are referable to the middle ear and mastoid due to the subsequent dysfunction of the eustachian tube.39,121 Individuals with Crouzon’s syndrome have a high incidence of jugular bulb dehiscence.122 The surgeon should be cautioned if this is diagnosed prior to myringotomy.
Cornelia de Lange syndrome is associated with cochlear hypoplasia (two turns) and ossicular deformity123
Internal Auditory Canal Anomaly
This is discussed more thoroughly in Chapter 8, but suffice it to say that on occasion, the imaging specialist may encounter IACs that are unusually wide, narrow, or perhaps even duplicated. Asymmetric canal widening is obviously associated with schwannoma. Dural ectasia (neurofibromatosis) may result in bilaterally large canals even in the absence of acoustic tumor (Fig. 5.40).124
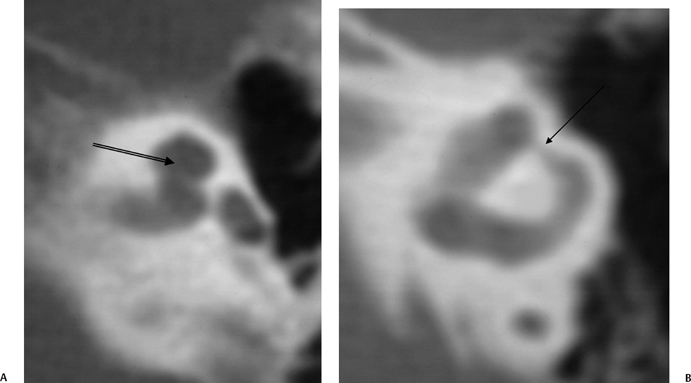
Fig. 5.37 Branchio-oto-renal syndrome. (A) Axial computed tomography (CT) of the left ear demonstrated modiolar deficiency (arrow). (B) Axial CT of the left ear reveals narrowing of the ampulla of the lateral semicircular canal (arrow). (Courtesy of Jan Casselman, MD.) (See Color Plate Fig. 5.37A.)
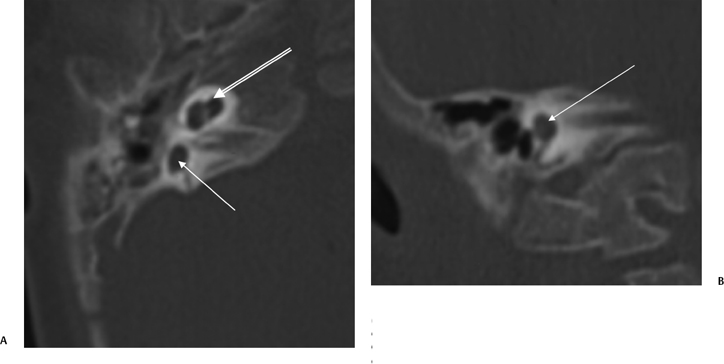
Fig. 5.39 CHARGE, semicircular canal (SCC) aplasia. (A) Axial and (B) coronal computed tomography (CT) images reveal complete absence of development of the SCCs, the classic finding in individuals with the CHARGE association. This patient also has some degree of modiolar deficiency (outlined arrow) and a small vestibule (thin white arrows).
There is a wide variation of the normal diameter of the IAC. Canals less than 2 mm in diameter are clearly abnormal.
Reduced IAC Caliber
Nonsyndromic congenitally narrow IAC is rare, especially as an isolated finding without additional inner ear anomalies, whereas acquired stenoses are more frequently reported, resulting from osteomas, exostoses, and fibrous dysplasia (see Chapter 8).125 The embryological nature of congenital IAC stenosis has not been completely elucidated, but most likely a developmental arrest of the vestibulocochlear nerve results in subsequent underdevelopment of the canal. This may be an isolated occurrence, or may it be associated with widespread labyrinthine deformity.
A severely hypoplastic IAC is classically associated with aplasia of the vestibulocochlear nerve (Fig. 5.41). In this circumstance, the canal transmits only the facial nerve, although dysfunction of each nerve in the canal can occur independently.125 Several reports have revealed that the size of the internal auditory canal in isolated cochlear nerve aplasia is variable and may be normal or only modestly decreased.126,127 There is better correlation with small size of the cochlear neural foramen (CNF) through which the cochlear nerve courses at the lateral end of the IAC. A stenotic CNF is virtually always accompanied by cochlear nerve deficiency (Fig. 5.42).
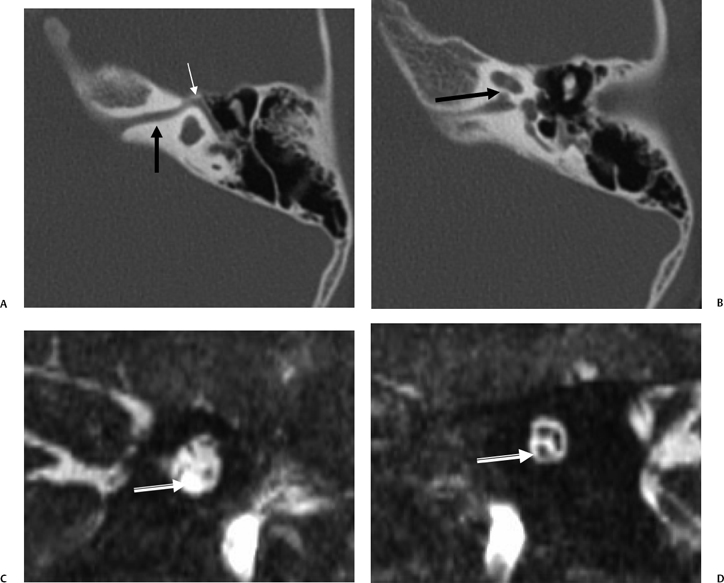
Cochlear nerve deficiency is not an uncommon cause of congenital hearing loss and can be reliably identified on thin-section T2WI sequences. There are three categories of cochlear nerve deficiency: type I—total absence; type IIa— absent or hypoplastic cochlear nerve, labyrinthine dysplasia; and type IIb—absent or hypoplastic cochlear nerve, morphologically normal labyrinth.128
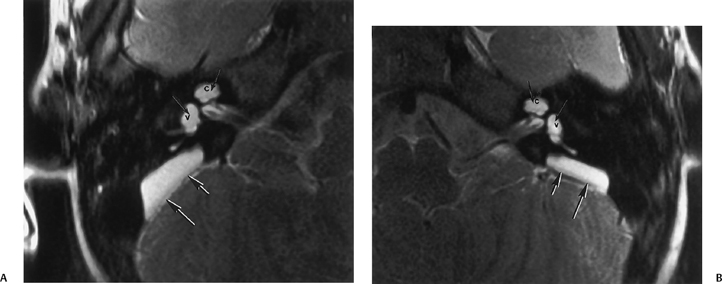
The integrity of the cochlear nerve is best seen on sagittal thin-section T2WI, which may reveal a small caliber cochlear nerve or even an entirely vacant anteroinferior quadrant at the fundus of the IAC. The caliber of the cochlear nerve is crucial information in patients who are candidates for cochlear implantation, as severe hypoplasia or absence is a contraindication to this procedure. As such, this entity is discussed further later in this chapter.
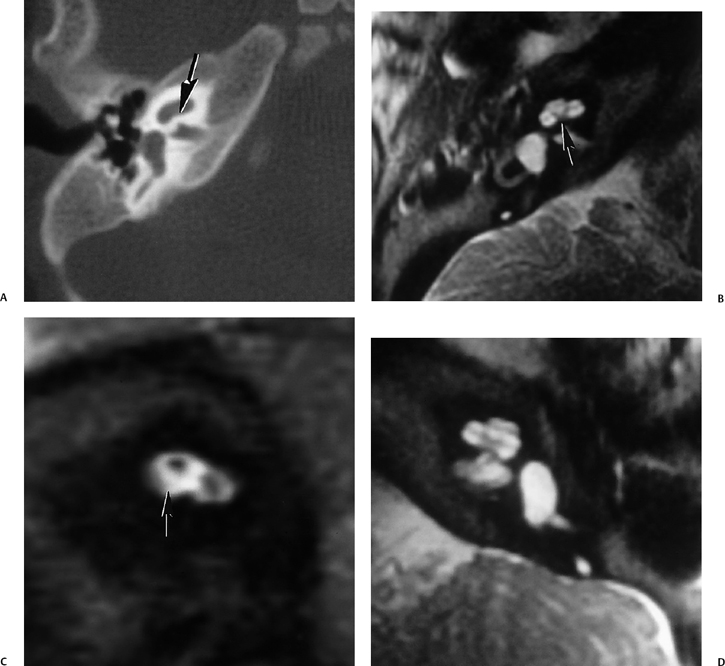
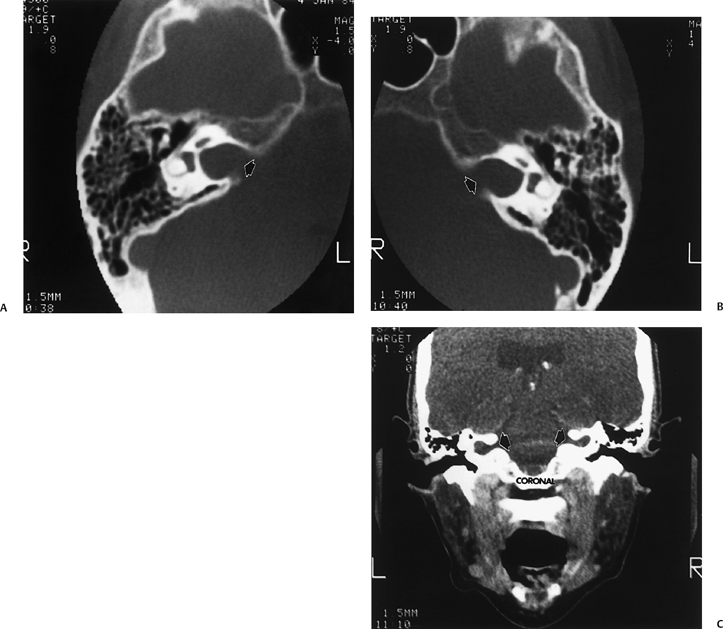
Fig. 5.42 Congenitally occluded cochlear neural aperture, cochlear nerve aplasia. (A) Axial computed tomography (CT), right ear. (B) Axial T2-weighted magnetic resonance image (T2WI), right ear. (C) Sagittal T2WI, right ear. (D) Axial T2WI, left ear. There is occlusion of the aperture through which the cochlear nerve normally travels (arrows, A,B). There is aplasia of the cochlear nerve, as indicated by the vacant anteroinferior quadrant of the internal auditory canal on sagittal T2WI through the fundus. This is a contraindication to cochlear implantation. Note the normal left ear for comparison.
X-Linked Progressive Mixed Deafness
In the patient with a wide IAC, careful study of the fundus and documentation of an intact bony partition between the IAC and the base of the cochlea and vestibule are crucial.129 Absence of the partition is associated with a strong potential for fistulous subarachnoid space–inner ear communication, increased inner ear pressure, and the propensity to develop a perilymphatic gusher upon stapes manipulation. A defective bony partition is often seen in association with other inner ear malformations or may be identified as an isolated finding. This latter circumstance has been documented to occur with an X-linked mode of inheritance (Fig. 5.22).130–134 These patients have a mixed hearing deficit, X-linked progressive mixed deafness (XLPMD) (Fig. 5.43 and Fig. 5.44).
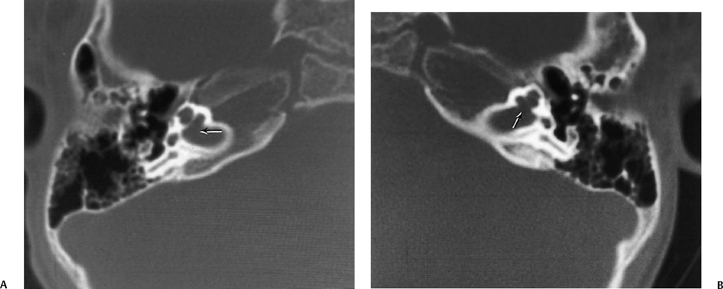
As indicated by the name of the disorder, hearing loss is mixed. The sensorineural component is due to increased pressure within the cochlear duct. Wide communication between the fundus of the internal auditory canal and the cochlear apex allows pressure from the subarachnoid space to be generated into the labyrinth, specifically the perilymphatic spaces (scala vestibuli and scala tympani) with subsequent impingement on the endolymphatic compartment (cochlear duct). The conductive component of the hearing loss is due to immobilization of the stapes. This immobilization is due to a higher than normal pressure of the perilymphatic compartment, specifically within the vestibule. This pressure interferes with movement of the stapes footplate and the transmission of sound waves.
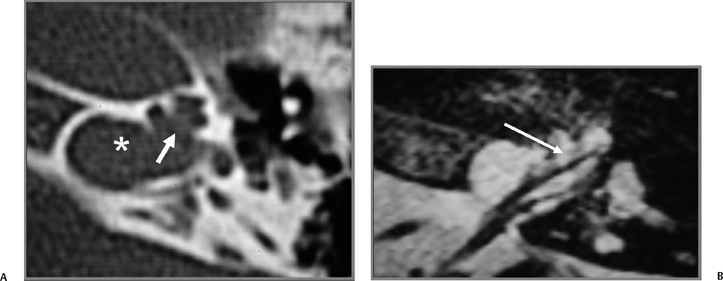
Fig. 5.44 X-linked progressive mixed deafness. (A) Axial computed tomography (CT) reveals a patulous internal auditory canal (IAC) (*) and a large defect at IAC fundus, resulting in direct communication with the cochlear apex (arrow). (B) Axial T2-weighted magnetic resonance image reveals normal caliber neural structures and confirms wide communication (arrow). (Courtesy of Jan Casselman, MD.)
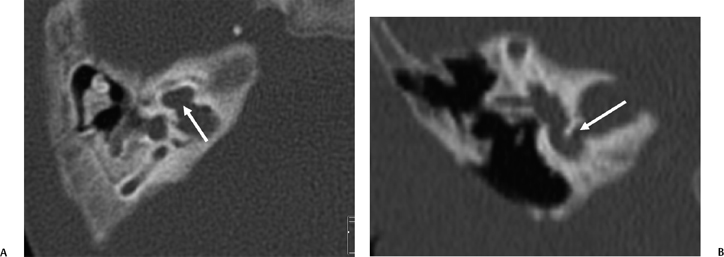
Related posts:
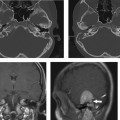
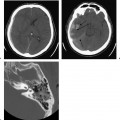
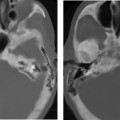
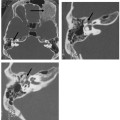
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


