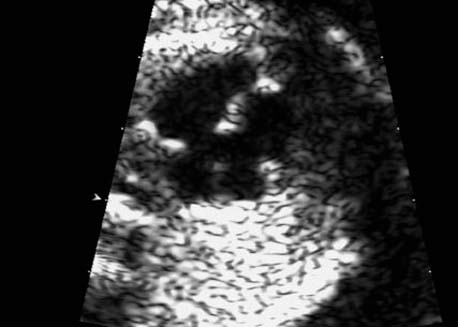
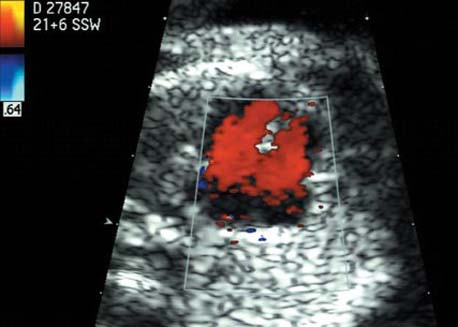
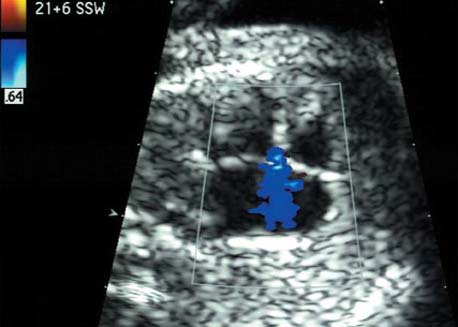
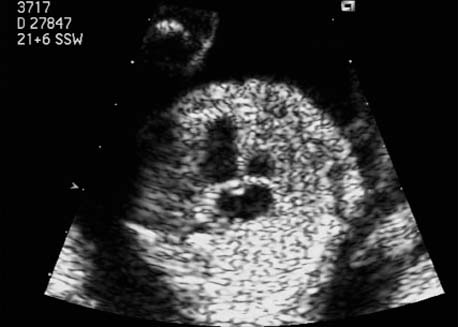
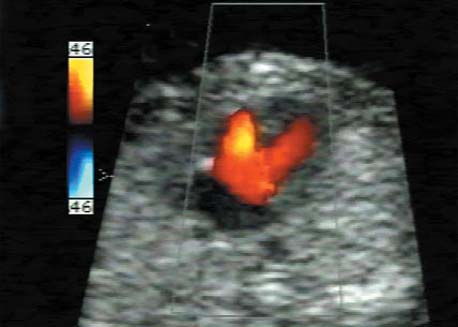
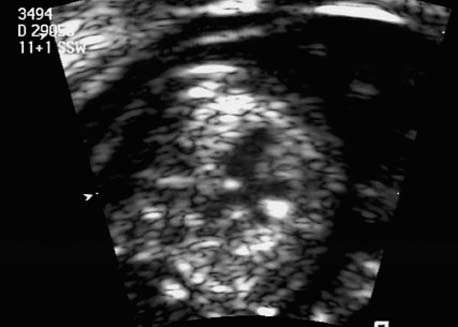
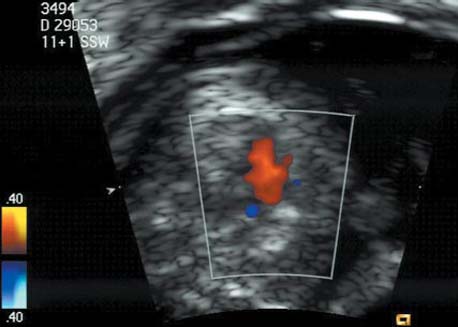
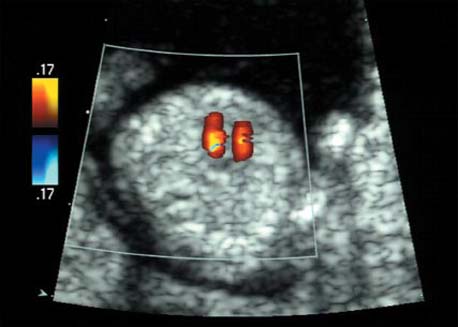
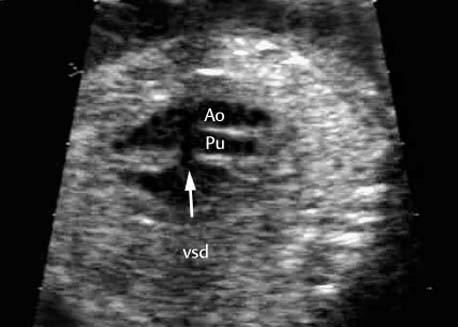
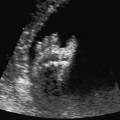
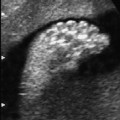
5 The Heart Congenital heart disease is relatively frequent, with an incidence of eight per 1000 live births (0.8%, or one affected child in 125 births). Half of these cases constitute severe cardiac anomalies, which require surgical intervention after birth. Not all malformations are detected prenatally, as some are due to the presence of the physiological fetal circulation between the heart and the great vessels (a normal state before birth) persisting even after birth; the malformations are therefore only detected after birth (e.g., open ductus arteriosus/Botallo duct). The rate of detection of cardiac anomalies during prenatal screening is extremely variable; according to the literature, it ranges from 5% to 85%, so that at best 17 of 20 cardiac anomalies may be detected prenatally, or at worst only one in 20. There are three reasons for this wide variation. 1 Time of Screening On the one hand, the frequency of cardiac malformations depends on the age of the pregnancy (cardiac anomalies which lead to spontaneous demise in early pregnancy are rarely detected at a later gestational stage). On the other hand, the sensitivity of the method of examination depends on the gestational age—i.e., the percentage in which the anomaly is detected. In our experience, the best time for cardiac screening is around 21 weeks. In obese patients, a control screening examination may further be necessary at 23 to 24 weeks. The present trend is to detect cardiac anomalies as early as possible; in individual cases, this can mean as early as 12 weeks. 2 Personal Competence “You can only detect what you know.” An experienced examiner who has seen a large number of cardiac malformations in pregnancy and who only carries out ultrasound examinations obviously has more certainty in his or her diagnosis and thus has a better chance of detecting an anomaly than an examiner who provides basic care and only carries out ultrasound examinations in pregnancy occasionally 3 Quality of the Equipment The quality of ultrasound equipment varies considerably, with new equipment costing anything between $20 000 and $200 000. In addition to B-mode sonography, color flow imaging Doppler sonography is a must for detecting fetal cardiac anomalies, as it establishes the direction of blood flow and determines flow velocities in such a way that detailed hemodynamic evaluation is possible. If cardiac anomaly is suspected after prenatal screening, then the following measures are taken: 1. In case of cardiac or any other anomalies, it is important to obtain a second opinion from another specialist in ultrasound screening to confirm the initially suspected diagnosis. 2. If the diagnosis of cardiac malformation is confirmed, then a detailed examination of the structures involved must be carried out. This is easier said than done, as it may take up to 2 hours in a physician’s practice to confirm the diagnosis with certainty and to decide what sort of consequences the anomalymay have. It is useful to involve a pediatric cardiologist at this stage who can interpret this and who can also counsel the patient. 3. Cardiac anomalies may be an isolated finding, may appear in association with other malformations, or may constitute part of a syndrome. Thus, it is absolutely necessary to do a complete and detailed fetal screening for other malformations to confirm either an isolated finding or a complex syndrome with an accompanying cardiac anomaly. 4. Cardiac malformation may indicate a chromosomal anomaly—for example, an AV septal defect is associated with trisomy 21 in about 50% of cases. After an anomaly of this type has been diagnosed, the parents should be informed about further tests that are available for detecting genetic abnormalities (amniocentesis and the prenatal Quick test, especially the fluorescent in-situ hybridization (FISH) test, placental biopsy, or fetal blood sampling). 6. Detecting a cardiac anomaly does not necessarily alter the plan of antenatal care or the method of delivery. Usually, a normal delivery can be expected at due date. However, the pregnancy must be monitored more often using ultrasonography. 7. The presence of a cardiac anomaly does not always mean delivery by cesarean section, but the delivery should take place in a center in which a pediatric intensive-care unit as well as an experienced pediatric cardiology team are available. After birth, the cardiologist performs an echocardiography and decides which other measures are immediately necessary. Anomalies such as ventricular septal defect or atrial septal defect do not always have to be treated, as many resolve spontaneously within the first year of life. The advantages of fetal echocardiography can be summarized as follows: 1. The main advantage of routine fetal echocardiography during pregnancy is to dispel any fear of having cardiac anomalies when the fetal ultrasound screening is normal. 2. If a severe cardiac anomaly is diagnosed or cardiac malformation suggests genetic disease, the parents have the option of whether or not to carry on with the pregnancy. 3. If the mother decides to carry on with the pregnancy after diagnosis of a fetal cardiac anomaly, the prenatal diagnosis allows optimal monitoring of the pregnancy, labor, delivery, and neonatal care of the child, which otherwise might have had a very poor chance of surviving, if any. Information for the Mother of an Affected Fetus Title: CHASER (Congenital Heart Anomalies—Support, Education, Resources) Description: Opportunity for parents of children born with heart defects to network with other parents with similar needs and concerns. Education on hospitalization, surgeries, medical treatments, etc. Newsletter, information and referral, phone support. Heart surgeons and facilities directory. Scope: National Founded: 1992 Address: 2112 N. Wilkins Rd., Swanton, OH 43558, United States Telephone: 419–825–5575 (day) Fax: 419–825–2880 E-mail: chaser@compuserve.com Definition: Narrowing of the aorta over a variable distance near the junction of ductus arteriosus (Botallo’s duct). Incidence: One in 1600 births. Sex ratio: M:F=1.5:1. Clinical history/genetics: Multifactorial inheritance; risk of recurrence with one affected child 2%, with two affected children 6%; if the mother is affected, then the risk of recurrence rises to 18%, in case of an affected father it is 5%. Teratogens: Diabetes mellitus, high doses of vitamin A. Associated malformations: Other cardiac anomalies such as VSD, valvular aortic stenosis, transposition of the great arteries. Associated syndromes: Turner syndrome (35% of affected children have coarctation of the aorta). This vessel anomaly has also been described in over 25 different syndromes. Ultrasound findings: Asymmetrical heart chambers with dilation of the right heart (atrium, ventricle, and pulmonary trunk dilation) may be the first indication. Narrowing of the aortic arch itself is difficult to demonstrate, as this often appears secondary to constriction of Botallo’s duct in the neonatal stage and is less obvious prior to this. Cross-sectional view of vena cava, aorta and pulmonary artery (three-vessel view) is the best method of demonstrating the relative narrowing of the aorta. In extreme cases, it resembles a hypoplastic left heart. In mild cases, intrauterine diagnosis is often very difficult. Clinical management: Further sonographic screening. Karyotyping. Ultrasound monitoring at regular intervals to detect increasing asymmetry of the ventricles. Vaginal delivery is possible. Procedure after birth: The decision on whether to undertake early therapeutic intervention depends on the severity of the existing lesion or of aortic stenosis appearing after the closure of Botallo’s duct. In severe cases, prostaglandin can be given as a prophylactic measure (inadequate liver and kidney perfusion, severe dysfunction of the left ventricle). Early surgical therapy or balloon angioplasty is indicated if signs of heart failure or inadequate perfusion of the periphery develop. In children without symptoms, the operation can be delayed until the age of 3–5 years. Prognosis: The prognosis is good after successful surgical treatment. When the lesion is isolated, the perioperative mortality is below 1%. In the presence of other anomalies, however, the mortality rate rises to 5–15%. Recurrence of stenosis is expected in 15–30% of patients. Beekman RH. Coarctation of the aorta. In: Emmanouilides GC, Allen HD, Riemenschneider TA, Gutgesell HP, editors. Moss and Adams heart disease in infants, children, and adolescents, including the fetus and young adult. Baltimore: Williams & Wilkins 1995: 1111–33. Benacerraf BR, Saltzman DH, Sanders SP. Sonographic sign suggesting the prenatal diagnosis of coarctation of the aorta. J Ultrasound Med 1989; 8: 65–9. Bronshtein M, Zimmer EZ. Early sonographic diagnosis of fetal small left heart ventricle with a normal proximal outlet tract: a medical dilemma. Prenat Diagn 1997; 17: 249–53. Bronshtein M, Zimmer EZ. Sonographic diagnosis of fetal coarctation of the aorta at 14–16 weeks of gestation. Ultrasound Obstet Gynecol 1998; 11: 254–7. Franklin O, Burch M, Manning N, Sleeman K, Gould S, Archer N. Prenatal diagnosis of coarctation of the aorta improves survival and reduces morbidity. Heart 2002; 87: 67–9. Nomiyama M, Ueda Y, Toyota Y, Kawano H. Fetal aortic isthmus growth and morphology in late gestation. Ultrasound Obstet Gynecol 2002; 19: 153–7. Wenstrom KD, Williamson RA, Hoover WW, Grant SS. Achondrogenesis type II (Langer–Saldino) in association with jugular lymphatic obstruction sequence. Prenat Diagn 1989; 9: 527–32. Definition: The most frequent form is complete AV canal, characterized by a large septal defect involving the atrial as well as the ventricular septa and the presence of a common atrioventricular valve. In the case of an incomplete AV canal, the AV valvular rims are separated and at least one anomaly of the AV valve is present (mostly a “cleft anterior mitral leaflet,” combined with an atrial septal defect of the foramen primum type and/or an inlet VSD). Incidence: 3.6 : 10 000; 2% of all congenital cardiac malformations. Clinical history/genetics: Down syndrome is confirmed in 50% of cases; in a further 10%, there is Ivemark syndrome (agenesis of the spleen or polysplenia). The rate of recurrence is 2.5% if one sibling is affected and 8% with two affected siblings In the case of an affected mother, the recurrence rate is 6%, and it is 1.5% if the father is affected. Associated malformations: Fallot-type pulmonary stenosis, fibromuscular subaortic stenosis, coarctation of the aorta; AV valve stenosis; anomalies of the spleen (Ivemark syndrome). Hydrops develops only in association with fetal cardiac arrhythmia. Associated syndromes: Down syndrome, Ivemark syndrome. Ultrasound findings: A combined atrial and ventricular septal defect in the mid-region of the AV valve (inlet VSD). The anterior and posterior AV valves are fused together, or lie much closer together than is normally expected. Using color flow Doppler imaging, AV valvular incompetence can often be detected. Disturbances in stimulus conduction may lead to cardiac arrhythmia (AV block, extrasystoles). Fig. 5.1 Complete atrioventricular septal defect. Apical view of the four chambers, at 21 + 6 weeks. The ventricular septum ends below the level of the valve, primarysep tum is missing, tricuspid and mitral valves are not separated: a complete atrioventricular septal defect. Fig. 5.2 Complete atrioventricular septal defect. Same case, diastolic phase, using color flow mapping. Blood is streaming through a common valve into both chambers. Fig. 5.3 Complete atrioventricular septal defect. Same case, systolic phase. Incompetence of the communicating atrioventricular valve, with regurgitation. Fig. 5.5 Atrioventricular septal defect, 14 + 5 weeks. Apical view of the four chambers using color flow mapping. Clinical management: Further fetal ultrasound screening. Karyotyping. Ultrasound monitoring at regular intervals for early detection of cardiac failure or fetal hydrops. Vaginal delivery is possible. Procedure after birth: Immediate postnatal complications are not to be expected in the absence of stenosis of great vessels. However, all children require surgical correction, which is usually carried out during the first year of life. Initial symptoms develop at the age of 4–6 weeks after birth (tachypnea, tachycardia). Prognosis: In most cases, it is possible to correct the defect surgically. The perioperative mortality is 5%. The prognosis becomes unfavorable if obstruction of the pulmonary artery is also present. Fig. 5.7 Atrioventricular septal defect. Same case. Demonstration of blood flow over the AV valve using color flow mapping. Fig. 5.8 For comparison, the flow pattern in a normal fetus at 12 weeks. Achiron R, Rotstein Z, Lipitz S, Mashiach S, Hegesh J. First-trimester diagnosis of fetal congenital heart disease by transvaginal ultrasonography. Obstet Gynecol 1994; 84: 69–72. Allan LD, Anderson RH, Cook AC. Atresia or absence of the left-sided atrioventricular connection in the fetus: echocardiographic diagnosis and outcome. Ultrasound Obstet Gynecol 1996; 8: 295–302. Areias JC, Matias A, Montenegro N, Brandao O. Early antenatal diagnosis of cardiac defects using transvaginal Doppler ultrasound: new perspectives? Diagn Ther 1998; 13: 111–14. Benacerraf BR, Gelman R, Frigoletto FDJ. Sonographic identification of second-trimester fetuses with Down’s syndrome. N Engl J Med 1987; 1371–6. Feldt RH, Porter CJ, Edwards WD, Puga FJ, Seward JB. Atrioventricular septal defects. In: In: Emmanouilides GC, Allen HD, Riemenschneider TA, Gutgesell HP editors. Moss and Adams heart disease in infants, children, and adolescents, including the fetus and young adult. Baltimore: Williams & Wilkins 1995: 704–24. Murphy DJ Jr. Atrioventricular canal defects. Curr Treat Options Cardiovasc Med 1999; 1: 323–34. Paladini D, Calabrb R, Palmieri S, Dandrea T. Prenatal diagnosis of congenital heart disease and fetal karyotyping. Obstet Gynecol 1993; 81: 679–82. Park JK, Taylor DK, Skeels M, Towner DR. Dilated coronary sinus in the fetus: misinterpretation as an atrioventricular canal defect. Ultrasound Obstet Gynecol 1997; 10: 126–9. Stoll C, Dott B, Alembik Y, Roth MP. Evaluation of routine prenatal ultrasound examination in detecting fetal chromosomal abnormalities in a lowrisk population. Hum Genet 1993; 91: 37–41. Tennstedt C, Chaoui R, Korner H, Dietel M. Spectrum of congenital heart defects and extracardiac malformations associated with chromosomal abnormalities: results of a seven year necropsy study. Heart 1999; 82: 34–9. Definition: Heart frequency below 100 beats per minute. Incidence: One in 20 000 births. Clinical history/genetics: 50% of cases are due to a complete AV block resulting from an autoimmune disease. The rate of recurrence is 8% if one child is affected. Teratogens: Not known. Embryology: Autoantibodies can disturb the cardiac stimulus conduction system. If structural malformation is the cause, then the atrioventricular septum is mostly involved. Associated malformations: In 50% of cases, complex cardiac anomalies are present such as AV canal, VSD, atrial isomerism, or abnormal venous flow. Fetal hydrops must be excluded. If there is sinus bradycardia, malformation of the central nervous system and fetal growth restriction should be looked for. Ultrasound findings: A very slow heartbeat is the first sign detected in B-mode. M-mode echocardiography is needed for further differentiation. In a complete AV block, the atria and ventricles beat regularly but are completely dissociated from one another. In sinus bradycardia, 1 : 1 conduction takes place from the atria to the ventricles. In case of atrial bigeminal rhythm, certain isolated beats are transferred to the ventricles and others are not. In second-degree AV block, 3: 1 or 2 : 1 conduction to the ventricles is possible. Clinical management: Further sonographic screening, anti-Rho, anti-La and antiphospholipid antibodies should be determined either to confirm or to exclude autoimmune disease. If there is structural malformation, karyotyping is advised. After 32 completed weeks, premature delivery is an option if cardiac insufficiency and hydrops develop. At earlier gestation, maternal therapy with steroids or plasmapheresis may be successfully administered. There is no consensus regarding the mode of delivery. “Fetal distress” is difficult to diagnose using cardiotocography during labor, and repeated fetal blood analysis is only valuable for a short period. Pulse oximetry may help in this situation. Cesarean section should be considered if hydrops develops and if the prognosis is not hopeless due to other cardiac anomalies. Procedure after birth: Intensive cardiac care is needed in the neonatal stage. If there is a bigeminal pulse and sinus bradycardia, therapy is usually not needed. In the presence of a complete AV block with a bradycardia of under 55 bpm or generalized hydrops, pacemaker installation should be considered. Prognosis: This depends on the cause of the bradycardia. Cases of bigeminal pulse have a good prognosis and the condition may disappear spontaneously. However, in 1% of cases, tachyarrhythmia may develop. A heart frequency of 80–100 bpm is found in sinus bradycardia; this may be sufficient for normal development of the child. The prognosis is poor if a complete AV block resulting from a complex malformation is diagnosed, or if the frequency is below 55 bpm. The fetus is not affected hemodynamically if there is a second-degree AV block. Beinder E, Grancay T, Menendez T, Singer H, Hofbeck M. Fetal sinus bradycardia and the long QT syndrome. Am J Obstet Gynecol 2001; 185: 743–7. Brucato A, Frassi M, Franceschini F, et al. Risk of congenital complete heart block in newborns of mothers with anti-Ro/SSA antibodies detected by counterimmunoelectrophoresis: a prospective study of 100 women. Arthritis Rheum 2001; 44: 1832–5. Carpenter RJJ, Strasburger JF, Garson AJ, Smith RT, Deter RL, Engelhardt HTJ. Fetal ventricular pacing for hydrops secondary to complete atrioventricular block. J Am Coll Cardiol 1986; 8: 1434–6. Fermont L, Batisse A, Le Bidois J. Prenatal cardiology: can we treat the fetal heart? Pediatrie (Bucur) 1992; 47: 339–45. Friedman DM, Rupel A, Glickstein J, Buyon JP. Congenital heart block in neonatal lupus: the pediatric cardiologist’s perspective [review]. Indian J Pediatr 2002; 69: 517–22. Mendoza GJ, Almeida O, Steinfeld L. Intermittent fetal bradycardia induced by midpregnancy fetal ultrasonographic study. Am J Obstet Gynecol 1989; 160: 1038–40. Nurnberg JH, Weng Y, Lange PE, Versmold H. [Transthoracic pacing in a very low birth weight infant with congenital complete atrioventricular block. Case report; in German.] Klin Pädiatr 2002; 214: 89–92. Silverman NH, Enderlein MA, Stanger P, Teitel DF, Hamann MA, Golbus MS. Recognition of fetal arrhythmias by echocardiography. JCU J Clin Ultrasound 1985; 13: 255–63. Wladimiroff JW, Stewart PA, Tonge HM. Fetal bradyarrhythmia: diagnosis and outcome. Prenat Dial 1988; 8: 53–7. Yaman C, Tulzer G, Arzt W, Tews G. Doppler ultrasound of the umbilical vein in fetal 3rd degree atrioventricular block. Ultraschall Med 1998; 19: 142–5. Definition: This is a complex cardiac anomaly in which both great vessels arise from the right ventricle. The left ventricle is emptied through a VSD to the right ventricle. Incidence: Rare; 1.5–2% of congenital heart disease. Sex ratio: M:F=1:1. Clinical history/genetics: Sporadic occurrence. With one affected sibling, the rate of recurrence is 3–4%. Chromosomal aberration is found in 10–15% of cases. Teratogens: Not known. Associated malformations: Tracheo-esophageal fistula. Associated syndromes: Trisomy 18. Ultrasound findings: Three different forms of DORV are recognized: 1. Side-by-side positioning of the great vessels
General Considerations
Coarctation of the Aorta
References
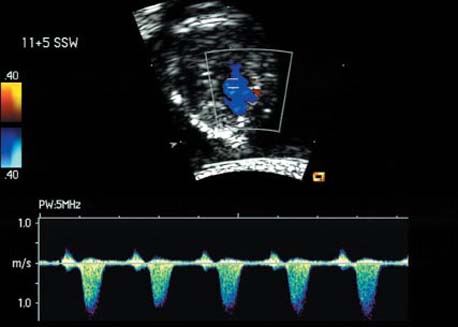
Atrioventricular Septal Defect (AV Canal)
References
Bradycardia
References
Double-Outlet Right Ventricle (DORV)
![]()
Stay updated, free articles. Join our Telegram channel


Full access? Get Clinical Tree


