Usual interstitial pneumonia (UIP) and idiopathic pulmonary fibrosis (IPF)
Nonspecific interstitial pneumonia (NSIP)
TABLE 13.1 Idiopathic Interstitial Pneumonias: Classification and Differential Diagnosis
Histologic Pattern
Idiopathic Clinical Syndrome
Differential Diagnosis of the Histologic Pattern
Usual interstitial pneumonia (UIP)
Idiopathic pulmonary fibrosis (IPF)
Collagen-vascular disease, asbestosis, drug toxicity, radiation, chronic hypersensitivity pneumonitis, familial pulmonary fibrosis
Nonspecific interstitial pneumonia (NSIP)
Nonspecific interstitial pneumonia (NSIP)
Collagen-vascular disease, hypersensitivity pneumonitis, drug toxicity, infection, immunodeficiency
Organizing pneumonia (OP); also known as bronchiolitis obliterans organizing pneumonia (BOOP)
Cryptogenic organizing pneumonia (COP)
Infection, bronchial obstruction, aspiration, drug reactions, collagen vascular disease, toxic-fume inhalation, radiation pneumonitis, hypersensitivity pneumonitis
Diffuse alveolar damage (DAD)
Acute interstitial pneumonia (AIP)
Acute respiratory distress syndrome (ARDS) of known cause (e.g., sepsis, shock, infection, drug toxicity, toxic inhalations, trauma)
Respiratory bronchiolitis (RB)
Respiratory bronchiolitis-interstitial lung disease (RB-ILD)
Smoking (usually), inhalations
Desquamative interstitial pneumonia (DIP)
Desquamative interstitial pneumonia (DIP)
Smoking (usually), inhalations
Lymphoid interstitial pneumonia (LIP)
Lymphoid interstitial pneumonia (LIP)
Collagen-vascular diseases (particularly Sjögren’s syndrome, rheumatoid arthritis, and lupus), immunologic disorders (e.g., Hashimoto’s thyroiditis, autoimmune hemolytic anemia, myasthenia gravis, pernicious anemia, chronic active hepatitis), infection, immunodeficiency (e.g., AIDS), drug toxicity
Cryptogenic organizing pneumonia (COP)
Acute interstitial pneumonia (AIP)
Respiratory bronchiolitis-interstitial lung disease (RB-ILD)
Desquamative interstitial pneumonia (DIP)
Lymphoid interstitial pneumonia (LIP)
helpful to think of the histologic patterns seen in the IIP as having a differential diagnosis, with one of the possible causes being an idiopathic syndrome (see Table 13-1). Since the radiographic appearances of the IIP parallel their histologic patterns, this approach allows the appropriate radiographic or high- resolution CT (HRCT) differential diagnosis to be suggested.
TABLE 13.2 Usual Interstitial Pneumonia (UIP) and Idiopathic Pulmonary Fibrosis (IPF): Histologic, Clinical, and HRCT Features | ||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||
A UIP pattern on HRCT or histopathology
The absence of an alternative etiology (e.g., collagen-vascular disease, asbestos exposure, drug treatment, hypersensitivity pneumonitis) after careful clinical evaluation (i.e., history, physical examination, pulmonary function testing, and laboratory assessment)
TABLE 13.3 Criteria for a Clinical Diagnosis of Idiopathic Pulmonary Fibrosis (2002) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
|
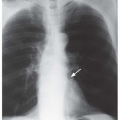
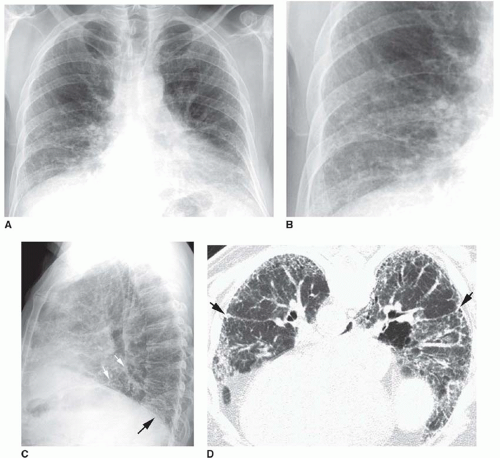 FIG. 13.1. Chest radiograph and HRCT in a patient with histologically proven idiopathic pulmonary fibrosis. A: PA radiograph shows reduced lung volumes. There is an increase in reticular opacities in the lung periphery and at the lung bases. The appearance and distribution are typical of idiopathic pulmonary fibrosis. B: Detail view of the right lower lobe shows increased reticular opacities. C: Lateral view shows increased reticular opacities in the posterior costophrenic angles (black arrow). A major fissure (white arrows) is bowed posteriorly because of more severe fibrosis in the lower lobe. D: Prone HRCT shows extensive subpleural reticular opacities with mild honeycombing. The major fissures (arrows) are displaced posteriorly because of lower lobe fibrosis. |
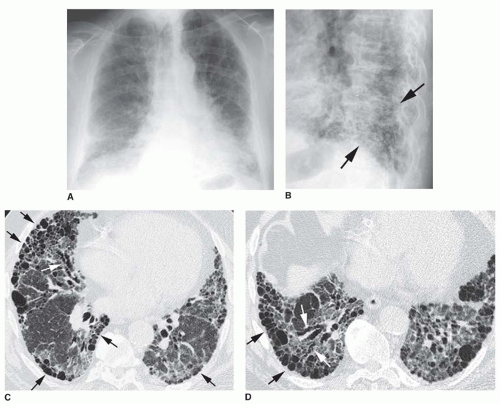 FIG. 13.2. Chest radiograph and HRCT in a patient with idiopathic pulmonary fibrosis. A: PA radiograph shows ill-defined reticular opacities at the lung bases. B: Coned-down lateral view shows a coarse reticular pattern (arrows) in the posterior costophrenic angles. C and D: HRCT shows extensive subpleural honeycombing (black arrows), reticular opacities, and traction bronchiectasis (white arrows). The honeycomb cysts range from a few millimeters to 2 cm in diameter. |
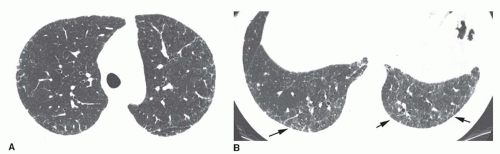 FIG. 13.3. Reticulation in early usual interstitial pneumonia/idiopathic pulmonary fibrosis (UIP/IPF). A and B: Concentric subpleural reticulation is the only abnormality. It is most severe in the lung bases (arrows B). Reticulation is the earliest abnormality seen on HRCT in patients with IPF. This appearance is nonspecific. Biopsy would be necessary for diagnosis. |
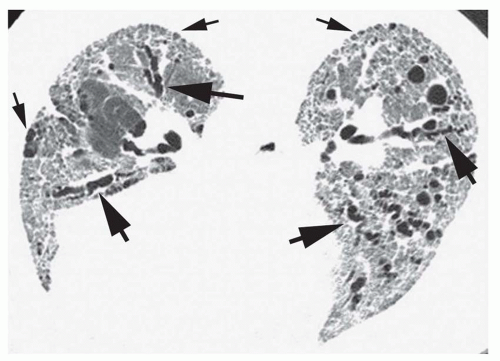 FIG. 13.4. Reticular opacities, traction bronchiectasis, and honeycombing in idiopathic pulmonary fibrosis. Prone HRCT shows a diffuse, coarse, reticular pattern at the lung bases, with traction bronchiectasis (large arrows) and mild subpleural honeycombing (small arrows). |
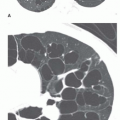
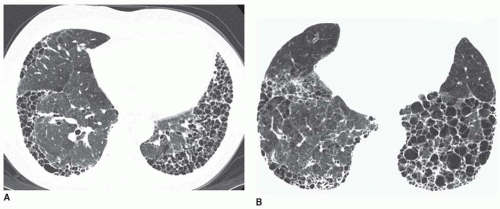 FIG. 13.5. Honeycombing on HRCT in two patients with idiopathic pulmonary fibrosis. A and B: Concentric subpleural honeycombing is visible in the lung bases. Most cysts are less than 2 cm in size. |
subpleural distribution. If these findings are present, the accuracy of HRCT in making the diagnosis of UIP exceeds 90%. In the absence of an associated disease (e.g., collagen-vascular disease) or exposure (e.g., asbestos, drugs, organic antigens), IPF is very likely the diagnosis. In patients with typical HRCT and clinical findings, lung biopsy is uncommonly performed.
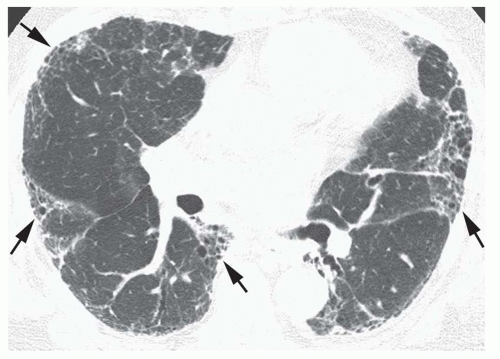 FIG. 13.6. Patchy subpleural reticular opacities and honeycombinga in idiopathic pulmonary fibrosis. Areas of honeycombing (arrows) involve the peripheral lung in a patchy fashion. |
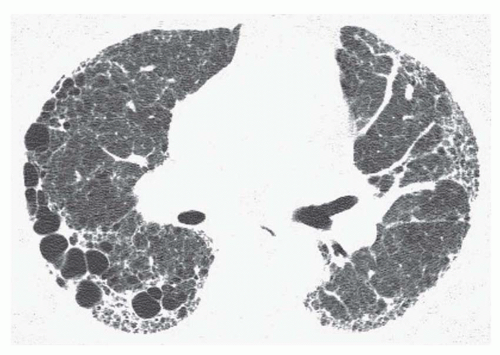 FIG. 13.7. Asymmetrical honeycombing in idiopathic pulmonary fibrosis. Subpleural reticulation and honeycombing are present. The honeycomb cysts are much larger on the right. |
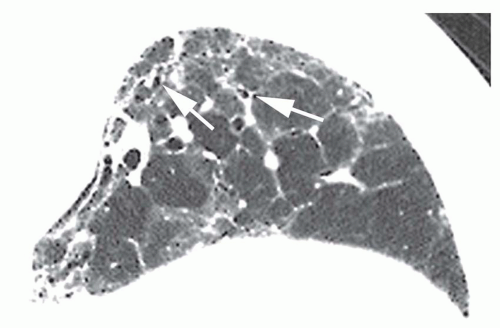 FIG. 13.8. Ground-glass opacity in idiopathic pulmonary fibrosis. Detail of the left lower lobe on a prone HRCT shows patchy ground-glass opacity. These regions also show irregular reticular opacities and traction bronchiectasis (arrows) and bronchiolectasis, indicating the presence of fibrosis. Ground-glass opacity is rare in idiopathic pulmonary fibrosis as an isolated finding but, as in this case, may be seen in association with reticular opacities. |
rate is 65% to 90%. Patients with NSIP may suffer an acute exacerbation similar to those occurring in UIP/IPF.
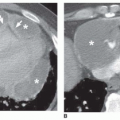
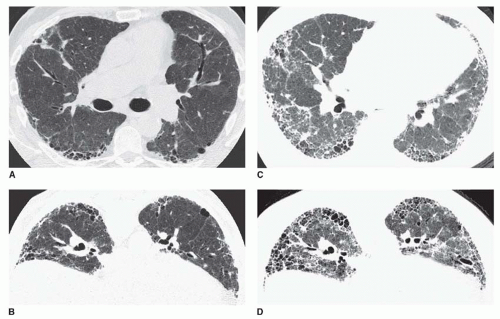 FIG. 13.9. Acute exacerbation of idiopathic pulmonary fibrosis. Supine (A) and prone (B) HRCTs show subpleural reticulation and honeycombing in the lower lobes, typical of idiopathic pulmonary fibrosis. Supine (C) and prone (D) HRCTs at the time of an acute exacerbation show progression of honeycombing and an increase in ground-glass opacity. This reflects diffuse alveolar damage. |
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


