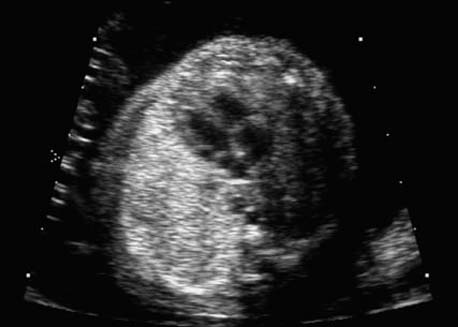
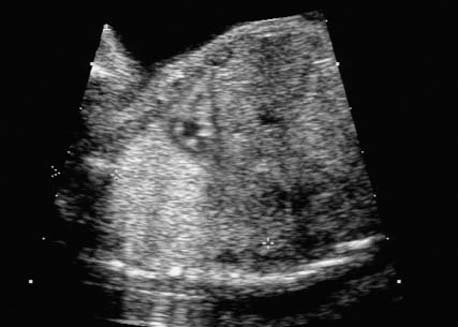
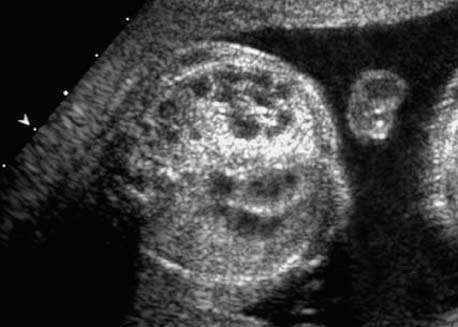
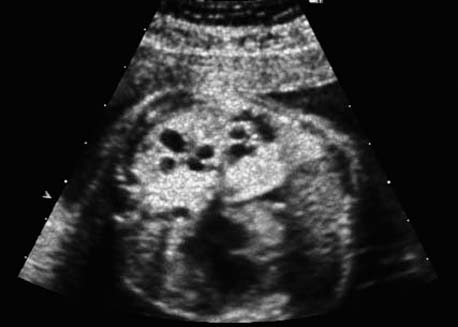
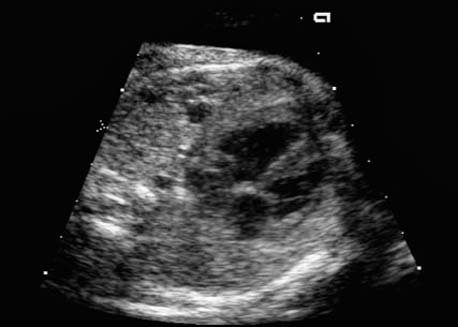
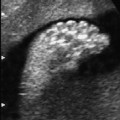
4 Thorax Definition: Congenital cystic adenomatoid malformation of the lung corresponds to a hamartoma with hyperplasia and dysplasia of the terminal bronchioli. Three forms are described (after Stocker): macrocystic form (type I), a mixed form (type II) and a microcystic form (type III). Incidence: Rare. Sex ratio: M:F=1:1. Clinical history/genetics: Sporadic occurrence. Teratogens: Unknown. Embryology: This malformation occurs in the first 6 weeks after conception due to dysplasia of the bronchioli (after conception). Associated malformations: These are found in 26% of cases, and consist of chest malformation (pectus excavatum), renal agenesis and fetal hydrops. Ultrasound findings: Stocker type I: Cysts measuring 2–10 cm in diameter; sometimes only one cyst is found. Stocker type II: Cysts smaller than 2 cm in diameter with echogenic tissue lying in between. Stocker type III: the echogenicity of the affected tissue is much stronger. In each case, displacement of the heart and the diaphragm is possible. Concomitant hydrops is found more often in type III. Up to sixty-five percent of cases develop hydramnios. Spontaneous regression of the lung anomaly has been known, such that the lesions may disappear completely. In addition, the echogenicity of the affected tissue may change during the course of pregnancy. Differential diagnosis: Diaphragmatic hernia, bronchogenic or neuroenteric cysts. Clinical management: Further ultrasound screening and regular monitoring, fetal echocardiography, if hydrops develops invasive treatment is necessary (cannulation of the cyst, drainage), or by reaching fetal maturity labor induction. Intrauterine surgery is still in experimental stages. Criteria for operative intervention (hydrops, size of the lesion, displacement of the heart) remain uncertain. Prophylactic treatment is disputed, as spontaneous resolution is possible. Procedure after birth: In severe cases, respiratory distress immediately after birth is common. Mechanical ventilation may even worsen the condition due to expansion of the lesion (over ventilation of nonfunctioning areas). Selective intubation of the healthy lung is a possibility. The newborn is placed on the side of the lesion to prevent overexpansion of the affected lung. The severity of the condition can be assessed postnatally using the magnetic resonance imaging (planning for surgery). The affected part of the lung is resected. Perioperatively, extracorporeal membrane oxygenation (ECMO) can be life-saving. Prognosis: The macrocystic form rarely causes fetal hydrops and has an overall survival rate of more than 70%. The microcystic form, associated with hydrops, has a poorer prognosis. If symptoms such as dyspnea and tachypnea appear in the newborn, then the mortality is up to 78%. In older studies, fifty percent of patients have clinical symptoms later in life, and have a good sure vival rate. In severe cases, resection of a complete lobe may be necessary, causing deformation of the thoracic cage. This can be corrected with placement of expanders. However, if part of the lung can be preserved, it expands, filling the hemithorax and resulting in improved functional capacity. Fig. 4.2 Congenital cystic adenomatoid malformation of the lung. Same patient as in the previous image, in a longitudinal section showing the liver and echogenic lung tissue. Fig. 4.3 Congenital cystic adenomatoid malformation of the lung. Left-sided anomaly at 21 + 2 weeks consisting of large cystic lesions, displacing the heart to the right. Fig. 4.5 Congenital cystic adenomatoid malformation of the lung. The same patient seen at 28 + 3 weeks. The findings are now less severe, with partial resolution of the echogenicity of the affected lung and displacement of the heart. References Adzick NS, Harrison MR, Glick PL, et al. Fetal cystic adenomatoid malformation: prenatal diagnosis and natural history. J Pediatr Surg 1985; 20: 483–8. Barret J, Chitayat D, Sermer M, et al. The prognostic factors in the prenatal diagnosis of the echogenic fetal lung. Prenat Diagn 1995; 15: 849–53. Bouchard S, Johnson MP, Flake AW et al. The EXIT procedure: experience and outcome in 31 cases. J Pediatr Surg 2002; 37: 418–26. Entezami M, Halis G, Waldschmidt J, Opri F, Runkel S. Congenital cystic adenomatoid malformation of the lung and fetal hydrops: a case with favourable outcome. Eur J Obstet Gynecol Reprod Biol 1998; 79: 99–101. Keidar S, Ben-Sira L, Weinberg M, Jaffa AJ, Silbiger A, Vinograd I. The postnatal management of congenital cystic adenomatoid malformation. Isr Med Assoc J 2001; 3: 258–61. Laberge JM, Flageole H, Pugash D, et al. Outcome of the prenatally diagnosed congenital cystic adenomatoid lung malformation: a Canadian experience. Fetal Diagn Ther 2001; 16: 178–86. Morris E, Constantine G, McHugo J. Cystic adenomatoid malformation of the lung: an obstetric and ultrasound perspective. Eur J Obstet Gynecol Reprod Biol 1991; 40: 11–5. Pinson CW, Harrison MW, Thornburg KL, Campbell JR. Importance of fetal fluid imbalance in congenital cystic adenomatoid malformation of the lung. Am J Surg 1992; 163: 510–4. Winters WD, Effmann EL, Nghiem HV, Nyberg DA. Congenital masses of the lung: changes in cross-sectional area during gestation. J Clin Ultrasound 1997; 25: 372–7. Zangwill BC, Stocker JT. Congenital cystic adenomatoid malformation within an extralobar pulmonary sequestration. Pediatr Pathol 1993; 13: 309–15. Definition: Esophageal atresia is a congenital disorder caused by closure of the esophagus with a blind end. In 90% of cases, there is an associated tracheo-esophageal fistula at the distal end. Incidence: One in 800–5000 births. Clinical history/genetics: sporadic occurrence. Teratogens: Retinoic acid, alcohol. Embryology: The anomaly arises due to defective division of the anterior intestines into trachea and esophagus during the 6th–7th week after menstruation (21–34 days after conception). In over 90% of cases, a tracheo-esophageal fistula is present, so that hydramnios—an important diagnostic clue—may be absent, as the amniotic fluid is diverted over the fistula into the stomach. Associated malformations: Other anomalies are evident in 50%, cardiac anomalies in 20%—especially ventricular septal defects (VSDs) or atrial septal defects (ASDs), other gastrointestinal defects in 10%, anal atresia in a further 10%, and VACTERL association in about 6%. Twenty-five syndromes have been described in association with esophageal atresia, with Down syndrome in 2–3%. Ultrasound findings: Esophageal atresia should be suspected when, in the presence of hydramnios (rare before 25 weeks), repeated screening examinations fail to demonstrate the fetal stomach. Due to the presence of tracheo-esophageal fistula in most cases, the stomach may be filled, but its small size is suspicious. In some cases, the blind end of the esophagus filled with fluid is detectable. Severe hydramnios may be present. In chromosomal anomaly or VACTERL association, fetal growth retardation may be evident. Failure to demonstrate the stomach in repeated examinations and concomitant hydramnios are suspicious signs for esophageal atresia. Clinical management:
Congenital Cystic Adenomatoid Malformation (CCAM) of the Lung
Esophageal Atresia
![]()
Stay updated, free articles. Join our Telegram channel


Full access? Get Clinical Tree


