Tumors of the Posterior Fossa and the Spinal Canal
Tumors of the Posterior Fossa and the Spinal Canal
Larry E. Kun
Shannon MacDonald
Nancy J. Tarbell
The posterior fossa occupies the lower half of the posterior cranial vault, bounded anteriorly by the clivus and posterior clinoid and inferiorly by the occipital bone and foramen magnum. Superiorly, the margin is defined by the tentorium cerebellae, that portion of the dura mater extending from the basisphenoid adjacent to the posterior clinoid, rising to cover the cerebellum, and extending posteriorly and inferiorly to insert at the level of the inion (the prominent midline outpouching of the occipital bone). The cerebellum and brainstem are located within the posterior fossa.
Nearly one half of all childhood brain tumors arise in the posterior fossa. The most common types are medulloblastoma, low-grade astrocytomas of the cerebellum, brainstem gliomas, and ependymomas (see Table 3.1) (1, 2, 3).
MEDULLOBLASTOMA
Medulloblastoma is a primitive cerebellar tumor of neuroectodermal origin. The tumor is the most common malignant brain tumor in children and adolescents, accounting for 20% of pediatric brain tumors or approximately 540 cases per year in the United States (1,3). Medulloblastoma was first identified in Bailey and Cushing’s 1925 classification of central nervous system (CNS) tumors (4,5). The classic description defined medulloblastoma as a primitive (embryonal) tumor of the cerebellum, derived from putative undifferentiated progenitor medulloblasts located in the cerebellar external granular layer.
The World Health Organization (WHO) classification of CNS neoplasms identifies embryonal tumors as a subset of the neuroepithelial neoplasms that are particularly prominent among pediatric brain tumors (6). The current WHO classification is included in Table 3.2, separately identifying the three major categories of embryonal CNS tumors: (1) medulloblastoma and its subtypes; (2) CNS primitive neuroectodermal tumors (PNETs, including supratentorial PNETs, CNS neuroblastoma, CNS ganglioneuroblastoma, medulloepithelioma, and ependymoblastoma); and (3) atypical teratoid/rhabdoid tumors. Note that pineoblastoma, considered clinically as a type of PNET, is listed separately as a tumor of the pineal region (6).
The most common embryonal CNS tumor is medulloblastoma, by definition a malignant embryonal tumor arising in the cerebellum, with predominantly neuronal differentiation. The tumor is thought to arise from cerebellar stem cells in the superficial external germinal layer (giving rise to cerebellar granule cells) or the deep-seated subventricular zone in the midline posterior medullary velum (generating cerebellar neuronal and glial cells) (7,8). DNA microarray gene expression patterns reported by Pomeroy et al. confirm the apparent derivation of medulloblastoma from the molecularly similar cerebellar granule cells, genetically distinct from supratentorial PNETs (9).
Histologically, medulloblastoma is a densely cellular neoplasm composed predominantly of undifferentiated small, round, blue cells. Differentiation may be toward neuronal or glial (astrocytic, oligodendroglial, and, less commonly, ependymal) lines in the more common “classic variant.” (6,10). Differentiation along mesenchymal lines (focal rhabdomyoblastic areas) define a variant called medullomyoblastoma (6,10). Approximately, 10% to 20% of medulloblastomas can be categorized as desmoplastic type, marked by relatively hypocellular areas of prominent nodularity in reticulin-free zones, occurring most often in the cerebellar hemispheres (6). Desmoplastic medulloblastoma is associated with mutations within the sonic hedgehog (SHH)-patched (PTCH) pathway and overexpression of IGF-2 (11, 12, 13). There is considerable excitement about the SHH pathway as a target for newly developing molecular-targeted therapies. Rodent models show the efficacy of cyclopamine in blocking the SHH pathway, resulting in dramatic shrinkage or prevention of the recombinant mouse medulloblastoma model (14,15). Anaplastic tumors are marked by nuclear pleomorphism and high mitotic rate; these tumors overlap with large cell medulloblastoma and are marked by chromosomal loss (17p-), MYC amplification, and poor prognosis (13, 16,17). Overexpression of ERBB2 may also be related to anaplastic large cell tumors and is a similarly negative prognosticator (12).
The histologic grade of medulloblastoma has only recently been linked to prognosis. Extensive nodularity has been correlated with favorable outcome; desmoplastic variant is similarly a marker of more favorable diseases (13,18,19). The degree of anaplasia (and, particularly, the subset of large cell anaplastic tumors) has been associated with inferior survival rates (13,16,17,20). Tumors with extraneural metastasis, either at diagnosis or as a pattern of failure, are more often associated with markedly anaplastic histology (18,21).
From the clinical genetics standpoint, medulloblastoma is the CNS tumor most often associated with germline mutations and familial diseases (see Table 3.3). The most frequent association is between Gorlin syndrome (nevoid basal cell carcinoma syndrome [NBCCS]) and desmoplastic medulloblastoma, both related to the tumor suppressor gene PTCH and the SHH receptor). In younger children, NBCCS is associated with extensive nodularity (19). NBCCS is characterized by somatic abnormalities (cutaneous nevi and palmar or plantar pits, odontogenic keratocysts of the jaw, bifid or fused ribs, falx calcifications) and development of numerous basal cell carcinomas, medulloblastoma, and rhabdomyosarcoma (12,19,22). In addition, mutations of the SHH-PTCH pathway are found in 10% to 20% of “sporadic” medulloblastoma (11,15). TP53 mutations mark the Li-Fraumeni syndrome, associated with a small percentage of medulloblastoma.
Mutations of the APC gene define Turcot syndrome of colonic polyposis, also seen in conjunction with medulloblastoma (6). Mutations of the WNT pathway, developmentally linked to proliferation of stem cells in the subventricular zone, were first noted in children with Turcot syndrome. The pathway is activated in 5% to 10% of sporadic medulloblastomas with classic histopathology, manifest by accumulation of intranuclear ß-catenin and associated with quite favorable prognosis; Wnt/Wg-active tumors are associated with isochromosome 16 (13,15,20, 23,24). Notch2 overexpression has also been noted in medulloblastoma, interesting as hypoxia appears to promote neural stem cell proliferation through Notch (25).
Other molecular correlations important in understanding the current directions in medulloblastoma include TrkC expression (directly proportional to survival in one major study) and ErbB2 expression. The latter factor is biologically related to cerebellar granular cell proliferation, migration, and invasion; elevated levels of ErbB2 are associated with poor outcome. (12,25,26)
The median age at diagnosis is 5 to 6 years. Approximately 20% of medulloblastomas present in infants younger than 2 years and 10% occur in young adults. Boys are affected more often than girls. Presenting symptoms are those classically associated with posterior fossa lesions in children: symptoms related to elevated intracranial pressure (headaches and vomiting, especially in the morning) and ataxia. Elevated intracranial pressure results from the tumor obstructing CSF flow through the sylvian aqueduct and the fourth ventricle.
Approximately 75% of medulloblastomas present in the midline cerebellar vermis. The tumor characteristically grows into and fills the fourth ventricle. Infiltration around the fourth ventricle is common, often involving the brachium pontis and extending onto the ventricular floor (i.e., the brainstem). Nearly one in four tumors arises within the cerebellar hemispheres, more commonly with desmoplastic histology. On magnetic resonance imaging (MRI), medulloblastomas are well-defined, typically solid lesions with uniform or, less often, nonhomogeneous contrast enhancement. Correlation between MR spectroscopic findings and metastasis at diagnosis has been reported (9). By computed tomography (CT) scan, the tumor often is hyperdense, reflecting high cellularity.
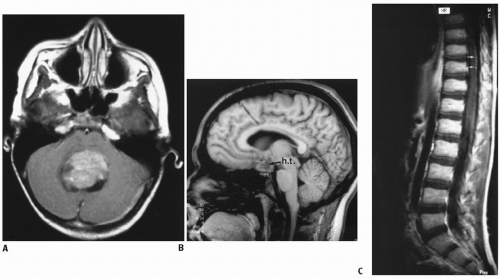
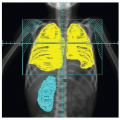
Medulloblastoma is the classic CNS tumor associated with CSF seeding or metastasis. The standard of care requires postoperative staging, typically based on imaging of the brain to assess degree of resection and potential subarachnoid metastasis (ideally within 24 hours, but acceptable up to 72-hours postsurgery), spinal MRI (gadolinium-enhanced study approximately 10 to 14 days after surgery to assess potential overt metastasis), and lumbar CSF cytology (best obtained immediately after spinal imaging). Subarachnoid dissemination has been reported at diagnosis in 20% to 35% of children (27,28). A review of 106 consecutive cases staged at diagnosis showed leptomeningeal disease in 32%, noted on both spinal MRI and CSF cytology in 12%, positive CSF alone in 8%, and MRI alone in 11% (20,28). Neuraxis disease typically involves the spinal subarachnoid space; intracranial metastasis is less common, noted as isolated disease in the basal or suprasellar cisterns (Fig. 4.1) or diffusely in the subarachnoid space (29).
Since publication by a radiation oncologist, the Chang (30) staging system has been used for clinical staging in medulloblastoma. The system was developed in the pre-CT era and is based on the size and invasiveness of the primary tumor at surgery (“T stage”) and evidence of spread outside the posterior fossa (“M stage”) (Table 4.1). Progressive tumor size and invasion of the brainstem defined increasing local tumor burden and aggressive behavior, classified as T1-4. With the advent of CT and MRI, it became apparent that imaging identification of brainstem invasion is not as reliable as surgical observation. There is no modern data to substantiate a role for T stage as an independent parameter predicting outcome or defining therapy (31, 32). Comparisons in otherwise early medulloblastoma (defined as M0 with complete or near total resection) and in series addressing advanced medulloblastoma have shown equivalent outcome among those with brainstem invasion (T3b) and those without such (T1-3a) (31, 33).
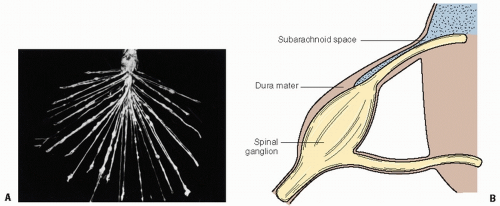
Figure 4.1 Medulloblastoma, originating classically in the cerebellar vermis (A,B), with signs of subarachnoid spread in the hypothalamic region (B) and along the spine (C).
No evidence of subarachnoid or hematogenous metastasis
T2
Tumor ≥3 cm in diameter
M1
Tumor cells found in cerebrospinal fluid
T3a
Tumor >3 cm in diameter with extension
M2
Intracranial tumor beyond primary site (e.g., into the aqueduct of Sylvius and/or into the subarachnoid space or in the third or foramen of Luschka or lateral ventricles)
Tumor >3 cm in diameter with unequivocal extension into the brainstem
M3
Gross nodular seeding in spinal subarachnoid space
T4
Tumor >3 cm in diameter with extension up past the aqueduct of Sylvius and/or down past the foramen magnum (i.e., beyond the posterior fossa)
M4
Metastasis outside the cerebrospinal axis (esp. bone marrow, bone)
aA pre-CT era system described by Chang (30), modified by J. Langston (personal communication, 1988).
b T3b is generally defined by intraoperative demonstration of tumor extension into the brainstem.
Note: T staging is of historical interest only, no longer used for clinical staging (33).
M stage is based on subarachnoid metastasis, progressively coding abnormal CSF cytology (M1) or imaging evidence of noncontiguous tumor in the cranium (M2) or spine (M3). Extraneural disease (most often confined to the bone marrow or bone) is present in fewer than 2% of cases at presentation, coded as M4. M stage remains a highly significant prognostic factor; intensity of therapy in current protocols and outcome are strongly related to the presence or absence of metastatic disease (31,32,34,35).
Current clinical trials and standard management in North America define clinical risk categories for medulloblastoma as average risk (children older than 3 years with no metastatic disease after near total or total resection, with less than 1.5 cm2 residual on early postoperative imaging) or high risk (overt metastatic disease based on CSF cytology or neuroimaging, or the presence of more than 1.5 cm2 residual on early postoperative imaging; more recently, all children younger than 3 years of age typically have been classified as high risk based on outcome studies) (36). With appropriately aggressive surgical intent in most centers in the United States and Europe, more than 65% to 75% of children above 3 years of age are staged as average risk. Of the 25% to 35% staged as high risk, more than 85% present with metastatic disease at diagnosis: primarily M3 (60%), but also M1 (30%), and M2 (10%); significant residual tumor at the primary site is present in >15% of cases (20,36,37).
Therapy
Surgery
Harvey Cushing’s (4) classic 1930 report of his experience with medulloblastoma demonstrated the inability of surgery alone to cure this tumor; only 1 of 61 patients survived 3 years after surgery with or without limited irradiation.
Maximal judicious surgical resection underlies most contemporary series (31,32). Gross total resection (i.e., no evidence of residual tumor seen at surgery and negative postoperative imaging) and near total resection (best defined as minimal residual: more than 90% resection estimated by the surgeon and less than 1.5 cm2 residual on postoperative imaging) have resulted in superior outcome in comparison to subtotal (51% to 90% resection) or partial (11% to 50% removal) resection and biopsy only (less than 10% removal) (32). Data from the Children’s Cancer Group (CCG) indicate gross total or near total resection in approximately 90% of children (31,32). Survival appears to correlate more significantly with amount of tumor residual (i.e., the surgical result as documented on immediate postoperative imaging) than with the surgeon’s impression of degree of resection; data confirming the value of minimal residual disease are most apparent among children with M0 disease (32,38). In an earlier CCG trial, 5-year event-free survival (EFS) was 78% for children with M0 disease and less than 1.5 cm2 residual, compared with 54% for those with larger residual volumes (31). For tumors adherent to or invading the brainstem, a report from St. Jude Children’s Research Hospital showed no advantage to pursuing gross total resection compared with near total removal, with none of the cases exhibiting more than 1.5 cm2 residual; morbidity appeared to be greater with the more aggressive surgical approach (39). With maximal safe resection a principle of therapy, the impact of minimal residual (<1.5 cm2) is difficult to discern; key is the distinct advantage of treatment on an average-risk regimen whenever possible, assuming such is a “given” for M0 disease (20,32,40, 41, 42).
Operative mortality has been reduced to 2% or less in pediatric neurosurgical centers. However, aggressive surgery may be associated with significant morbidity (32,43,44). The posterior fossa syndrome has been described in 15% to 25% of children after posterior fossa craniotomy (21, 45). The syndrome is signified by difficulty swallowing, truncal ataxia, mutism, and, less often, respiratory failure; recent imaging data suggest the etiology may be a cerebellocerebral diaschisis (45). Symptoms and signs typically are noted after a 12- to 24-hour period of initially uneventful postoperative recovery. Disabling neurologic signs often improve dramatically, sometimes over many months after surgery. It is important to maintain an aggressive, curative approach (including radiation therapy) in children with this syndrome, anticipating significant neurologic recovery, which may not be apparent early in the course of irradiation.
Table 4.2 Medulloblastoma-Results of Postoperative Radiation Therapy (No Chemotherapy)
CSI, Craniospinal irradiation; CrI, cranial irradiation; SpI, spinal irradiation; r, converted to rad; Gy, Gray; PFS, progression-free survival; OS, overall survival; M-SFOP, French Society for Pediatric Oncology, Medulloblastoma; PF, posterior fossa; fx, fraction.
The routine use of ventriculoperitoneal shunts to reduce intracranial pressure before posterior fossa craniotomy resulted in significant improvement in operative morbidity and mortality, when introduced 40 years ago. Children with ventriculoperitoneal shunts typically become shunt dependent. Shunt failure or infection may complicate long-term survival, necessitating revision or replacement in nearly 25% of children measured 5 years after insertion. In many academic pediatric neurosurgical centers, it is a standard procedure to place a ventricular drain (ventriculostomy), as needed, at the time of surgery. The surgeon often can document reestablishment of CSF flow after fourth ventricular tumor resection. Later shunt insertion may be needed in 20% to 25% of children (32,46,47). A delayed shunt insertion approach provides physiologic CSF dynamics for the majority of children, avoiding potential late events related to an indwelling ventriculoperitoneal shunt.
Radiation Therapy
The efficacy of radiation therapy in medulloblastoma was reported within a decade of Cushing’s initial description of the tumor. Cutler et al. (48) reported the radiation responsiveness of medulloblastoma and the value of preventive irradiation of the entire neuraxis based on Cushing’s clinical series. The seminal report documenting cure of medulloblastoma with craniospinal irradiation (CSI) was published by Bloom et al. in 1969 (49), documenting 32% survival at 5 years and 25% disease-free survival at 10 years. Numerous reports have subsequently confirmed increasing rates of disease control with modern radiation techniques; at standard CSI dose levels, radiation therapy alone achieves durable disease control in 65% to 75% of patients with average-risk disease (Table 4.2) (33,50). Modifications of radiation volume, dosage, and fractionation have been explored. The outcome following postoperative irradiation alone in average-risk medulloblastoma using conventional radiation parameters (Pediatric Oncology Group [POG]-CCG trial, one arm of which used CSI to 36 Gy, posterior fossa boost to 54 Gy resulting in 65% 7-year EFS in a large cohort evaluated largely by CT myelography rather than spinal MRI scans) has been used as a basis for nonrandomized comparisons in establishing current standards for combined modality therapy in North America. The result is systematic reduction in CSI dosage (to 23.4 Gy; v.i.) with well-documented efficacy now in average-risk disease when combined with contemporary cis-platinum-based chemotherapy (20,51, 52, 53). Agreement on combined chemoradiation is based on disease control rates that appear to be superior to those achieved with irradiation alone for both average-risk and highrisk presentations, a randomized European trial demonstrating improved outcome with chemoradiation compared to contemporary radiation therapy alone, and several studies suggesting improvement in the risk:benefit ratio based on dose-volume modeling and evolving clinical data (52,54, 55, 56, 57, 58).
Chemotherapy
Phase II trials have documented the chemoresponsiveness of medulloblastoma to alkylating agents (cyclophosphamide, CCNU[lomustine]), platinum compounds (cisplatin, carboplatin), etoposide (administered orally or intravenously), antimetabolites (methotrexate), and camptothecins (topotecan) (59, 60, 61, 62).
The sentinel trial documenting the efficacy of adjuvant chemotherapy was reported by CCG, combining the attenuated CSI dose in average-risk patients that had just shown only 55% EFS at 5 years in the POG-CCG trial referenced in the prior section with concurrent vincristine and postirradiation cis-platinum, vincristine, and CCNU; the 79% progression-free survival (PFS) at 5 years confirmed earlier institutional experience to show among the best disease control rates then documented in this disease (33,34,51). The International Society for Pediatric Oncology (SIOP)/United Kingdom Children’s Cancer Study Group (UKCCSG) PNET-3 trial showed improved EFS with limited preirradiation chemotherapy and full-dose irradiation versus equivalent irradiation alone: 78% EFS at 5 years with preirradiation vincristine, etoposide, carboplatin, and cyclophosphamide compared to 65% with irradiation alone (54). A large randomized trial assessing reduced-dose CSI followed by cisplatin and vincristine with “standard” CCNU versus cyclophosphamide confirmed overall EFS more than 80% with no difference in disease control on either chemotherapy arm; early analysis suggests a larger number of secondary neoplasms may be apparent in the cyclophosphamide arm (53). St. Jude reported a sizable prospective trial using postirradiation “compressed” cyclophosphamide, vincristine, and cisplatin; 83% EFS was obtained without sometimes toxic vincristine during irradiation and with a marked reduction in ototoxicity attending postirradiation cisplatin when the latter was given with amifostine (20,63). The standard of care for children with average-risk medulloblastoma throughout North America has been accepted as reduced-dose CSI (23.4 Gy) followed by chemotherapy including an alkylating agent, vincristine, and cisplatin (Table 4.3).
For patients with high-risk disease, studies through the 1990s typically showed 5-year EFS at the 40% to 50% level following full-dose irradiation and chemotherapy (41,42). St. Jude’s SJMB96 study has shown 70% 5-year EFS following the same compressed chemotherapy noted above, preceded by full-dose CSI (20). Randomized trials have shown somewhat conflicting results regarding the sequence of postoperative therapy: POG showed >60% 5-year EFS in high-risk medulloblastoma regardless of postoperative/preirradiation chemotherapy (cyclophosphamide, vincristine, cisplatin) or the opposite sequence, both using full-dose CSI (64). The German HIT’91 trial showed superior results with postoperative irradiation followed by CCNU/vincristine/cisplatin compared to postoperative ifosfamide/etoposide/high-dose methotrexate/cisplatin cytosine arabinoside followed by irradiation: 83% 5-year EFS compared to 53%, respectively for M0 patients; no difference was noted in the M2-3 cohort, both at <40% EFS (42). CCG 9931 documented a 17% PD rate during a prolonged, 5-month preirradiation regimen, again showing only 43% EFS in highrisk disease (58). Similar trials have noted that outcome in average-risk patients receiving preirradiation chemotherapy correlates with response to chemotherapy; in the Milan trial, those with CR/PR to preirradiation chemotherapy enjoyed 94% PFS compared to 61% if only SD or PD attended chemotherapy (65). Several studies note the time to initiating irradiation is conversely related to disease control (41,47).
For disease recurrent after radiation therapy (with or without chemotherapy), numerous studies demonstrate chemotherapy responsiveness to single agents, multiagent combinations, and high-dose therapy with hematologic stem cell rescue. Except in the infant setting, best defined as “radiation therapy naïve,” durable secondary disease control following initial CSI has only rarely been achieved despite aggressive, high-dose chemotherapy and further irradiation (66, 67, 68, 69, 70). Local irradiation can provide further control at the primary site (71). Trials of intrathecal chemotherapy in this setting are of interest, but to date with only limited phases I and II data (72).
Radiotherapeutic Management
Volume
Medulloblastoma is the seminal tumor identified with subarachnoid dissemination. The need for full CSI has been recognized for more than five decades. In reviewing serial treatment regimens in Sweden, Landberg et al. (73) noted serial improvements in survival rates with increasing radiation volume: 5% 10-year survival after limited posterior fossa irradiation, 15% after irradiation to the posterior fossa and spinal canal, and 53% after CSI. Reported failures in the subfrontal region additionally indicate the need to completely encompass the cranial and spinal subarachnoid spaces; such failures clearly represent inadequate dosage to the subfrontal area, a potential site of geographic miss even in the era of 3D, imaging-based treatment planning (74, 75, 76, 77). Other parameters key to appropriate CSI encompassing the entire subarachnoid volume are discussed below (see Techniques, p. 61-64).
Prospective trials preceding the current decade have been based on the standard use of boost irradiation to the entire posterior fossa. Use of 3D conformal radiation therapy (3D CRT) to the entire posterior fossa provided some reduction in cochlear dose, important in children receiving cisplatin and at-risk for both chemotherapy- and radiationrelated ototoxicity (Table 4.4) (57,63,78, 79, 80, 81). The use of 3D CRT targeting only the tumor bed has been shown to be equivalent to treatment encompassing the entire posterior fossa, with in-field or posterior fossa recurrences at or below the 5% level even with more limited boost volumes (40,52,57,82). Initial experience with a 2 cm anatomically defined clinical target volume (CTV) expansion of the gross tumor volume (GTV) (postoperative tumor bed) showed few if any posterior fossa failures outside the targeted volume (78,83,84). The St. Jude SJMB96 trial confirmed local disease control in 95% of cases with the 2-cm CTV; the recent M-SFOP 98 and current COG trials are testing a 1.5-cm CTV expansion, while a previous study from the University of Washington and the current SJMB03 study have studied a 1 cm anatomically defined expansion to identify the CTV (40,52,82).
Detailed reviews of actual dosimetry and dose modeling, including 3D CRT, intensity-modulated radiation therapy (IMRT), and proton beam radiation therapy demonstrate significant dose reduction to the cochlea and upper cervical spinal cord with tumor bed targeting and 3D-planned therapy; more narrowly defining the target volume and more conformal treatment offer the potential to significantly spare the medial temporal lobes (and, potentially, the hippocampus, both volumes related to cognitive function) in addition to anterior cranial structures (optic chiasm, hypothalamus) (52,57,85).
Although the earlier literature had suggested a predominance of posterior fossa failures in medulloblastoma despite surgery and relatively high-dose irradiation to the entire posterior fossa using broad 2D techniques, more recent studies consistently indicate a shift in the pattern of failure toward diffuse leptomeningeal recurrences; failures limited to the posterior fossa occur in less than 5% to 10% of patients (33,51,78,83,84,86,87). Together with the several trials reporting outcome after local boost to the tumor bed, recent experience supports a major prospective trial of image-guided irradiation to limited tumor bed boost volumes, as planned for 2004 in COG.
Dosage
Medulloblastoma is a radiosensitive tumor. In vitro studies by Fertil and Malaise (88) demonstrated a favorable surviving fraction in vitro at 2 Gy (28%), comparable to most other embryonal pediatric tumors and notably different from the clinically less responsive malignant gliomas. Earlier data indicated a correlation between dosage to the primary tumor site and the outcome; local disease control and survival were consistently related to posterior fossa dosages of ≥50 to 55 Gy using conventional fractionation at 160 to 180 cGy per fraction (46,86,89). The use of limited volume boost to 59.4 Gy for patients with imaging evidence of residual disease at the primary site has been reported, with no clear evidence that escalation beyond 55 Gy is fruitful (78,84).
Table 4.3 Major Medulloblastomas Studies Reporting Outcome with Postoperative Irradiation Combined with Chemotherapy
Recent trials support the use of 23.4 Gy (at 180 cGy/fraction) to the neuraxis for average-risk presentations when combined with effective chemotherapy as discussed above. Largely in response to age-related concerns regarding neurocognitive changes, a small pilot study of 18-Gy CSI for 10 children younger than 10 years was undertaken at the Children’s Hospital of Philadelphia and at the University of Pennsylvania. Disease control in seven of ten children was initially reported with apparently only minor neuropsychologic deficits (90). Subsequent reports suggest failures following the 18-Gy level may be more apparent with time (91). The current COG aver-age-risk medulloblastoma protocol randomizes children with average-risk disease between 3- and 8-years-old to 23.4-Gy CSI or 18-Gy CSI, prospectively addressing the potential efficacy and relative toxicities of further reduction in CSI dosage.
For patients with high-risk disease, particularly those with overt metastasis (including M1 in most studies in addition to M2-3), 36 Gy to the neuraxis remains the standard. Several studies have used 39.6 to 40 Gy for cases with significant bulk disease through the intracranial and spinal meninges (20).
Trials of hyperfractionated CSI include neuraxis dosages of 30 to 48.4 Gy at 100 to 110 cGy per fraction twice daily, with an interfraction interval greater than 6 hours; cumulative dosages for the posterior fossa have been 66 to 72 Gy with similar fractionation (58,82,92,93). Long-term results of the French M-SFOP 98 trial showed 75% 6-year PFS in average-risk medulloblastoma after hyperfractionated CSI (36 Gy in 36 fractions) with tumor bed boost (cumulative dosage, 68 Gy in 68 fractions); the goal with equivalent total CSI dose for aver-age-risk disease treated with irradiation alone was to reduce toxicity while preserving equivalent disease control (50,82). A concurrent Milan study used preirradiation chemotherapy and hyperfractionated accelerated radiation therapy for M+ disease, with 1.36 Gy b.i.d. to 31.2 to 39 Gy (depending on age and response to chemotherapy), with 1.5 Gy b.i.d. to the posterior fossa to cumulative levels of 59 to 60 Gy; at investigator’s option, cases with overt primary residual or progressive tumor received additional treatment to a reduced target volume to 9 Gy; preliminary reports seem favorable albeit without details regarding long-term toxicities to date (65). The potential value of altered fractionation has yet to be demonstrated.
Technique
The goal of achieving uniform dosage throughout the subarachnoid space, encompassing the entire intracranial vault and spinal canal, is one of the more technically demanding aspects of radiation oncology. Several techniques are appropriate for CSI administration (94). The fundamental is the use of opposed lateral fields including the cranium and upper cervical spinal canal, matching a posterior spinal field including the full spinal subarachnoid space or, in larger children, the upper one half of the spinal canal (with a separate, matched lower posterior spinal field) (Figs. 4.2 and 4.3).
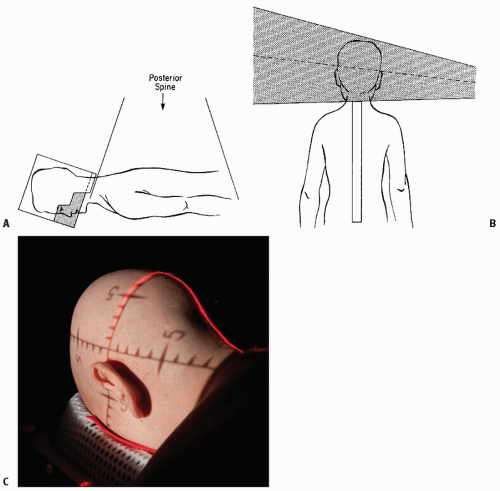
It is important to establish immobilization and reproducibility of setup. Prone positioning is preferable because it allows greater immobilization and better extension of the chin (minimizing potential bone growth changes caused by the exit of the posterior spinal field) and simplifies technical maneuvers at the critical junction between the lateral craniocervical fields and the posterior spinal field(s). Immobilization can be achieved through a customized plaster cast, use of the Alpha Cradle system, or a vacuum bag (discussed in Chapter 22). CT simulation for CSI provides both accuracy and flexibility in aligning fields and junction zones (95). Supine positioning may occasionally be medically necessary; detailed attention to the junction area is needed when one adapts the more familiar prone geometry to supine CSI use (96, 97, 98).
Figure 4.2 Consideration of spinal volume for craniospinal irradiation. A: Nerve root seeding in medulloblastoma; this figure first appeared in the Bulletin of the Los Angeles Neurological Society (8:1-10, 1943), representing advanced, diffuse leptomeningeal seeding. B: Typical anatomy of the nerve roots demonstrating the extension of the subarachnoid space laterally along the nerve root to a location in the neural foramen.
Figure 4.3 Craniospinal irradiation (CSI). Basic technique for CSI including lateral cranial volume (A) and appropriately matched posterior spinal field(s) (B). C: Prone positioning with Aquaplast mask.
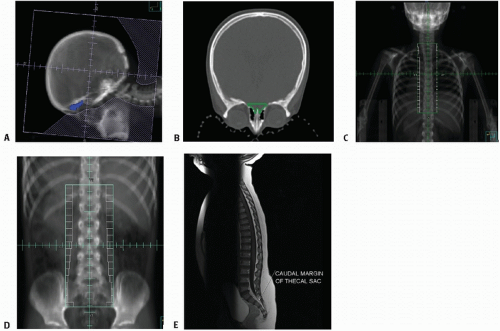
Figure 4.4 Craniospinal irradiation (CSI). A: Lateral cranial volume defined to encompass the subarachnoid space, with attention to the cribriform plate (blue), shown in (B). C: Posterior spinal volume, here divided for upper (thoracic) spine with lateral margins to dosimetrically include the neural foramina. D: Posterior spinal volume for lower (lumbosacral) spine, including “flair” to encompass sacral neural foramina (see Fig. 4.2). E: MRI depicting lower thecal sac.
CT simulation provides axial and sagittal anatomy to enable one to accurately outline the key anatomic sites and plan rational junction areas (95). Key areas, already discussed, include attention to the cribriform plate—outlining that structure on axial images greatly facilitates accurate identification of target volumes at the “tight” interface of the cribriform plate and the eyes (76) (Fig. 4.4). Other critical anatomic sites important in designing full cranial volumes include the middle cranial fossa and a sufficient margin about the calvarium to be certain to achieve full-dose levels within. In younger children, the margin above the eye may be extremely close but should permit coverage of the subfrontal area as a first priority, minimizing dosage to the eye and, in particular, the optic lens. For older children, the pneumatized frontal sinuses allow sufficient margin to obviate concern for the cribriform anatomy (Fig. 4.4). The literature supports the use of customized blocking rather than the multileaf collimator (MLC) to ensure adequate subfrontal coverage while minimizing dosage to the eye (99); new MLC technology with ≤5-mm leaf size over larger fields may permit MLC blocking in the near future.
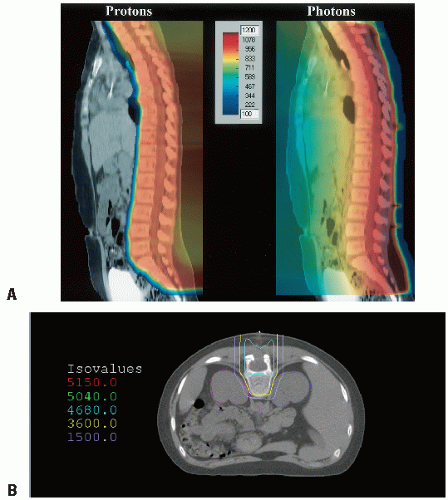
Defining the lower border of the spinal field should always be based on the lowest level of the thecal sac as determined by MRI, typically at or below S2 (Figs. 4.4E and 4.5) (100,101). The width of the spinal field should be sufficient to dosimetrically encompass the full width of the spinal canal to the dorsal nerve roots laterally. Adequate lateral coverage at the lower sacral margin is appropriate to cover the sacral foramina (Fig. 4.2) (101). Several institutions have reported the relative advantage in using intensity-modulated photon irradiation or, more recently, proton beam approaches (Fig. 4.5) (102,103). Proton spinal irradiation significantly spares the underlying heart, breasts, and soft tissues; long-term follow-up is not available as yet documenting late effects (102,104, 105, 106). Homogeneity in planning and delivering CSI with protons is challenging; introduction of scanning beam technology seems to provide more optimal dosimetry (Fig. 4.6) (107).
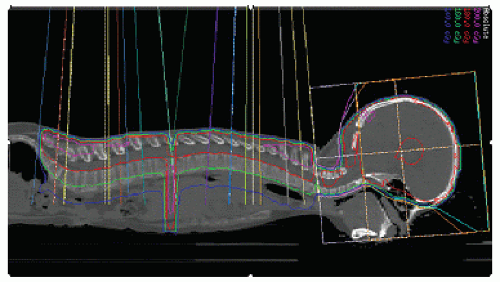
Figure 4.5 Craniospinal irradiation (3). Use of multiple “IMRT minibeam” compensates for diameters in lateral cranial volumes and depths in posterior spinal volumes to provide relatively homogeneous dose distribution for photon CSI. Note lateral dosimetric projection confirms adequacy of lower thecal sac coverage (see Fig. 4.4A).
Figure 4.6 Spinal irradiation. A: Comparison of dosimetry with single PA field, PBRT versus photon. B: PBRT dosimetry closely conforming to spinal canal and vertebral body (to ensure relatively “normal” growth).
The brain and upper cervical spine are treated with lateral fields. The collimators for the lateral fields are angled to match the divergence of the posterior spinal field (Fig. 4.3). The cranial fields must accommodate serial decreases in length to feather the junction zone; discrete readjustments of the junction by 5 to 10 mm/week are typical, if there is some literature regarding daily modulation of the junction (108). A table angle generally is used to correct for caudal divergence of the craniocervical fields (Fig. 4.3). Although detailed analysis shows only minimal inhomogeneity if a correcting table angle is not used (94), full 3D attention at the junction zone permits more confident abutting of the lateral craniocervical and posterior spinal fields. Even with the ideal craniocervical match using asymmetric jaws, it is important to feather all junctional zones, shifting the anatomic junction site by at least 5 mm every 8 to 9 Gy, effectively once a week (109). Helical tomotherapy has been used, with excellent handling of the complex, full neuraxis (110).
Figure 4.7 Tumor bed boost for resected medulloblastoma. GTV = tumor bed (green) with 1-cm anatomic 3D expansion to provide protocol-specified (SJMB03) CTV (blue) and further 3-mm geometric expansion to establish PTV (red). Shown are transverse planes (A = MRI, B = CT at level of cochlea, shown in blue) and sagittal plane (C = MRI in midline showing spinal cord, brainstem, chiasm in yellow, and pituitary in green).
The cranial fields include the entire intracranial subarachnoid space. The most difficult area is at the level of the cribriform plate, where there is often little margin to allow one to block the eyes and dosimetrically encompass the critical subfrontal cribriform site (76). Even with 3D proton therapy, oblique beam configurations are required to reduce incidental lens dose (111). For CSI, there are both dosimetrically modeled data and direct clinical measures to document the relative advantage of the classic Cerrubend delivered by parallel-opposed or obliqued lateral fields. Current boost techniques typically include either (1) 3D CRT or IMRT to encompass the posterior fossa, or (2) using similar techniques to encompass the tumor bed alone, using anatomically corrected 3D expansion by 1 to 2 cm to define the CTV (40,52,57,82, 84,112). With photon irradiation, the differences between IMRT or 3D CRT and conventionally planned 2D therapy are impressive in relatively sparing the cochlea, temporal lobes, and brainstem (11,40,52,57). The current COG trial in average-risk medulloblastoma randomizes all cohorts between 3D-based therapy to the posterior fossa or to the tumor bed (using a 1.5-cm anatomic expansion → CTV and a 3- to 5-mm geometric expansion → PTV (COG: ACNS0331). In nonprotocol settings, there is now sufficient information to suggest the adequacy of boost volumes confined to the tumor bed (52,57,82,84). This sophisticated approach entails analyzing the initial tumor extent based upon pre-operative MRI (or CT) and identifying margins of the posterior fossa or the post-operatively reconfigured posterior fossa tumor bed using CT and, ideally, fused pre-operative MR imaging. (Fig. 4.7 demonstrates boost confined to the tumor bed). The goal of image-guided irradiation and narrow margins in targeting the tumor bed is to diminish the dosage to the cochlea (especially important with combined irradiation and cisplatin), the temporal lobes, and the hypothalamic-pituitary region (Fig. 4.7). Several reports document the theoretical advantage of proton beam therapy for the “boost” volume, based on significant further reduction in normal tissue doses to the key anatomic regions noted: cochlea, temporal lobes, and pituitary-hypothalamic region (11, 104, 113).
The quality of radiation therapy has been correlated with improved outcome, based on both volume adequacy (in several but not all prospective cooperative group reviews) and duration of therapy (with several reports indicating inferior disease control when the interval to – complete irradiation exceeds 45 days) (46,54,86,114, 115, 116).
Delivery
CSI results in predictable, if quantitatively variable, acute changes in the peripheral blood counts. Monitoring for neutropenia or thrombocytopenia, most often noted during or after the third week of CSI without preceding chemotherapy, is critically important. Traditionally, CSI is interrupted if absolute neutrophil count falls below 1000 cells per milliliter, especially if the child is febrile. When necessary to permit completion of neuraxis irradiation, granulocyte colony-stimulating factor may be used to correct neutropenia; thrombocytopenia may necessitate platelet transfusion (117). Irradiation is initiated with CSI. If blood counts necessitate delaying or interrupting CSI, initiation of posterior fossa irradiation will provide continuity and limit unnecessary interruption in irradiation at the primary site. One should return to CSI as soon as hematologic status permits. With preirradiation chemotherapy regimens, further delaying the start of radiation therapy can negatively impact the outcome (41, 47,54).
Nausea and vomiting are generally more pronounced in older children. Use of antiemetics is important in preventing “anticipatory” vomiting, which may be more difficult to control. Ondansetron usually is successful. Rarely, corticosteroids may be necessary at low dosage levels, particularly early in the postoperative period. Nutritional support during CSI is mandatory as children often go from completion of irradiation to adjuvant chemotherapy.
Results
Long-term disease-free survival after surgery and irradiation should now approach the 70% level overall (Table 4.2). Factors associated with more favorable outcome include age greater than 3 years, localized presentations (i.e., M0), and tumors amenable to near total or total resection. The more effective combinations of radiation therapy and chemotherapy for children older than 3 years show long-term disease control in 80% of those with average-risk disease, and 60% to 70% of those with high-risk medulloblastoma (Table 4.3) (20,50,53,65). The impact of histology, biologic findings, and genetic analyses is increasingly important, as summarized above. Investigations to additionally improve the outcome seek improvement in disease control and reduction in functional limitations attendant to therapy.
EMBRYONAL AND MALIGNANT GLIAL TUMORS IN INFANTS AND YOUNG CHILDREN
Children younger than 3 years account for 15% to 25% of pediatric CNS neoplasms (1,118,119). Symptoms in this age group usually include enlarged head, lethargy, and vomiting. Tumors are predominantly supratentorial; in comparison to older children, infant tumors are more often malignant and may be more frequently metastatic at diagnosis (120). The most common tumor types include astroglial tumors (primarily low grade; among infants < 1-year-old, up to 25% are high-grade malignant gliomas), the embryonal neoplasms (medulloblastoma and the supratentorial embryonal tumors, including PNETs and pineoblastomas), and ependymomas (121, 122, 123). Atypical teratoid/rhabdoid tumors (AT/RTs) occur predominantly in this age group (6,124,125). A significant proportion of intracranial teratomas and choroid plexus tumors present in young children below 12 to 18 months of age (119,122). Infantile desmoplastic neuroepithelial tumors (desmoplastic infantile gangliogliomas and astrocytomas) also arise predominantly in the very young. These lesions often are quite large, are peripherally located, and appear aggressive histologically, but typically display rather “benign,” low-grade behavior, rarely recurring after primary resection (6,126,127).
Survival rates for the embryonal brain tumors presenting in children younger than 3 to 4 years are lower than for older children (122,123,128,129). Tumor type, pattern of growth, and the therapeutic ratio for both surgery and radiation therapy are unfavorable when compared to older children (32,130,131). Operative morbidity and mortality rates are higher in infants than in older children; after radiation therapy, cognitive dysfunction, somatic alterations, endocrine deficits, and neurotoxicity are more pronounced than in older children (55,56,122,132).
For malignant gliomas, there is actually suggestion that outcome exceeds that of older children and adults, based on apparent differences in biology and disease response to chemotherapy (133, 134, 135, 136).
Therapy
For embryonal tumors with long-established chemosensitivity (particularly medulloblastoma, but also supratentorial PNETs), a number of trials between 1985 and 2000 explored the use of prolonged primary postoperative chemotherapy using delayed, diminished, or no irradiation (depending on the goals and philosophy of the group or institution) (Table 4.5). Several large series documented a high rate of chemoresponsiveness to a “standard” four-drug regimen (including cyclophosphamide, cisplatin, vincristine, etoposide) or to systemic methotrexate; durable disease control without irradiation was limited to 25% to 35% of cases in most trials, typically in those with localized disease amenable to complete resection at diagnosis (120,122,123,132,137, 138, 139, 140, 141, 142, 143).
Two different directions have proven more successful, at least in selected settings, over the past 10 to 15 years. Successive trials from the German POG tested progressively more intense systemic and intrathecal methotrexate with an alternating drug program incorporating the agents noted above. While overall PFS in the HIT SKK 87 trial (1987 to 1993) was 53% in the favorable resected, M0 cohort (with identical overall survival [OS]), the study showed youngsters with desmoplastic medulloblastoma enjoyed nearly 90% PFS. The SKK 92 study (1992 to 1997) intensified methotrexate and noted overall 5-year PFS of 58%; among the resected M0 group, 5-year PFS was 82% (with 14 of 17 survivors treated with surgery and chemotherapy only, absent irradiation which was used only for residual/progressive disease during chemotherapy). Once again, the results with desmoplastic histology were exceptional: 85% PFS (95% OS) compared to 34% PFS (41% OS) in those with classic medulloblastoma (142,144).
The second direction was suggested by Khalifa and the French group (SFOP), where primary chemotherapy showed only 29% PFS at 5 years even among the most favorable, resected M0 cohort. Notable was the OS rate of 73%, reflecting excellent “salvage” therapy with high-dose chemotherapy, busulfan-thiotepa, and local irradiation; among 39 patients treated, 5-year postsecondary treatment survival was 77% for those with M0 disease initially and at failure (140,145). Although the St. Jude group had also documented excellent salvage with CSI alone, the functional consequences of more limited irradiation in this age group seem “axiomatic” (132,138). Both POG and the Pediatric Brain Tumor Consortium (PBTC) initiated trials in the late 1990s testing chemotherapy (including intrathecal mafosfamide, an activated form of cyclophosphamide, in the PBTC study) with planned, localized irradiation after the initial 4 months of chemotherapy. Results are yet in analysis, recognizing that among the M0 group that proceeded to consolidative local irradiation on PBTC 001, 5-year PFS is 85% and OS, 95% (Kun L, Blaney S. 2009, Personal Communication).
All infant trials to date have shown poor outcome for the 20% of patients presenting with neuraxis dissemination, OS rates rarely exceeding 10% to 25% (139,140, 142,144). Although CSI is curative in a significant proportion of such children, the consequences of CSI at effective dose levels are not considered acceptable (132). Alternative use of aggressive, high-dose chemotherapy alone has been fraught with otherwise unseen toxicity (including toxic deaths) and EFS for favorable (M0, resected) presentations approximating 50%; outcome in the M+ cohort has been essentially zero (141).
Separate from medulloblastoma is the immature, highly aggressive AT/RT (6,146). AT/RTs occur predominantly in young children, presenting in the posterior fossa; those occurring in children older than 3 years are more often supratentorial lesions (129,146,147). The lesions are histologically distinctive, and diagnosis by light microscopy and immunohistochemistry (especially documenting positive epithelial membrane antigen and vimentin) is often definitive. The tumor is associated with monosomy of chromosome 22, a finding in common with extraneural primary rhabdoid tumors (148). Genetically, the tumor is associated with loss of the tumor suppressor gene hSNF5/INI1 in more than 75% of cases; absence of INI1 by FISH is diagnostic (129,147,149). Tumor that histologically might be called infant medulloblastoma or other embryonal diagnoses with documented loss of INI1 is considered AT/RT and should be handled as such. Up to 15% to 25% of cases show leptomeningeal dissemination at diagnosis (149,150). Although AT/RTs often respond to chemotherapy (especially carboplatin-containing regimens), the disease course has been marked by rapid recurrence and neuraxis dissemination (146,147). There is an increasing evidence that the outcome is related to postoperative irradiation; recent trials incorporate early local irradiation for children as young as 12 to 18 months old, ideally limiting postoperative chemotherapy to 4 to 6 weeks (125,129,147, 151). For children older than 3 years of age, use of postoperative CSI followed by chemotherapy has resulted in 78% 2-year EFS compared to 11% for younger children in whom irradiation was delayed or avoided (129).
Surgery
As in older children, complete resection is often the primary predictor of disease control; for infant medulloblastoma, the differences in outcome strongly favor attempted GTR in every major series regardless of the type and intensity of postoperative management (120,122,123,139, 140, 141,144). In the initial Baby POG study, OS for medulloblastoma was 40%, compared with 60% for the one third of children who had undergone GTR and 69% for those with GTR and localized disease (120). In the latest published GHOP trial, PFS among all M0 cases falls from 82% to 50% based on the absence or presence of residual tumor postsurgery, respectively (144).
Delayed definitive surgery has been utilized for sizable medulloblastomas or supratentorial PNETs in this age group. After initial chemotherapy, tumors may be reduced in size and vascularity, resulting in more successful tumor resection (123,138).
Choroid plexus tumors are often malignant carcinomas in this age group. The tumors typically arise in the lateral ventricles; histology can be uncertain in predicting benign or malignant behavior, with carcinomas marked largely by brain invasiveness and atypia. Complete resection alone appears to be adequate therapy, with few recurrent tumors following imaging-confirmed removal even without added chemotherapy or irradiation (152, 153, 154).
Radiation Therapy
Evolving combinations of systematic or selected consolidative irradiation, “standard” irradiation for disease progression, or multimodality salvage regimens incorporating lowor high-dose radiation therapy have resulted in radiation therapy as a component of therapy for nearly half of all surviving children in this admittedly “vulnerable” age group (120,132,138,140,141,145). Important in the context of current strategies is identification of those cases most likely to benefit from local irradiation, with consensus developing toward noting those with classic (nondesmoplastic) histology and localized medulloblastoma or those with incompletely resected M0 desmoplastic medulloblastoma. Using “early” planned irradiation, typically within the first 4 months of postoperative chemotherapy, is key to avoiding the scenario of requiring more aggressive irradiation (volume and dose) and chemotherapy (dose) for those who progress during or after more prolonged chemotherapy. In AT/RT, even earlier initiation of radiation therapy (4 to 8 weeks) is key to improving outcome, as noted above.
Although salvage CSI (at therapeutic dosage levels of 30 to 36 Gy using 180-cGy daily fractions) has been successful in controlling more than 40% of recurrent medulloblastomas (in fact, equally effective in M0 and M+ cases in earlier St. Jude data), the ultimate 40% to 60% disease control was balanced by a median IQ of only 62 at 7 years (132). The latter finding has dampened enthusiasm for salvage CSI, at least at dosage levels greater than 24 Gy, in this age group.
Only gold members can continue reading. Log In or Register to continue