Fig. 4.1
Induction of glycolysis by HIF. Stabilization of the transcription factor HIF in low oxygen conditions leads to the transcription of a large number of genes that encode for proteins involved in promoting the glycolytic pathway as shown in the figure
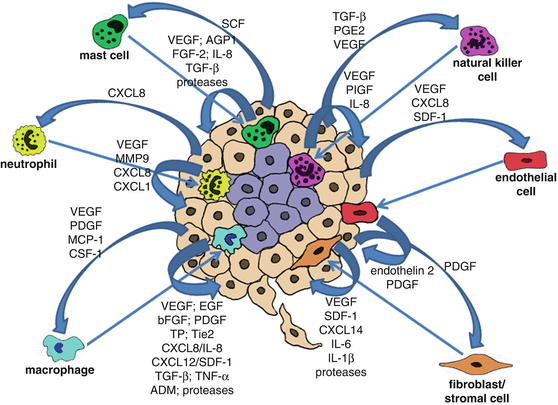
Fig. 4.2
The role of paracrine signalling between cancer cells and tumour associated cells (microenvironment) in tumour angiogenesis . Cancer cells secrete proteins that function as chemoattractants to tumour associated cells, such as macrophages, neutrophils, mast cells, natural killer cells, endothelial cells and fibroblasts/stromal cells. Recruited tumour associated cells in their turn secrete proteins that will further stimulate cancer cell growth/proliferation, tumour angiogenesis and recruitment of cells to the tumour site
4.4 Hypoxia Induced Tumour Angiogenesis
Another critical response, essential for tumour survival under hypoxic conditions is the formation of new blood vessels, which will provide oxygen and nutrients that are essential for tumour survival and growth, a process known as tumour angiogenesis .
HIF induces the transcription of vascular endothelial growth factor (VEGF), platelet derived growth factor (PDGF), angiopoietin and eritropoietin genes that are involved in the promotion of angiogenesis [9, 15–18]. VEGF is particularly important in tumour angiogenesis, being highly secreted not only by cancer cells, but also by tumour associated cells such as macrophages and other immune cells, as well as cancer associated fibroblasts (CAFs) (reviewed in detail bellow). VEGF binds to the VEGF receptor (VEGFR) at the surface of endothelial cells which constitute the internal layer of the blood vessels, stimulating in this way endothelial cell proliferation, survival, secretion of matrix degradation enzymes (e.g. matrix metalloproteases and plasmin) and migration to the tumour site [9].
4.5 The VEGF Family of Pro-angiogenic Proteins
Taken into account the complexity of the process of angiogenesis (described in detail bellow), it is remarkable that a single growth factor, VEGF, regulates this process so predominantly. The human genome contains five genes encoding for distinct VEGF family members, namely VEGF (also called VEGF-A), placenta growth factor (PlGF), VEGF-B, VEGF-C and VEGF-D. Structurally, the VEGF family of proteins are homodimers, constituted by two subunits of about 120–200 amino acids in length [19]. The VEGF family distinguishes itself from other angiogenic protein families by the fact that its members have largely non-redundant functions. VEGF is the main component of this family, and it stimulates angiogenesis both in physiological and pathological processes by signalling through the VEGF receptor-2 (VEGFR-2, also known as FLK1) [20, 21]. In contrast to VEGF, PlGF and VEGF-B appear to have a relatively minor role in the regulation of angiogenesis, but have been shown to play a role in cardiac muscle function [22, 23]. VEGF-C, a ligand of the VEGFR-2 and VEGFR-3 receptors, activates blood-vessel tip cells [24, 25]. VEGFR-3 activation by VEGF-C has been shown to lead to the formation of blood vessels during early embryogenesis, but later becomes a key regulator of lymphatic angiogenesis – the formation of new lymphatic vessels from pre-existing vasculature [26]. VEGF-D binds to VEGFR-3 and is also involved in lymphatic angiogenesis [24].
4.6 The Mechanism of Angiogenesis
In the developing mammalian embryo, angioblasts differentiate into endothelial cells, which assemble into a vascular labyrinth, a process known as vasculogenesis. Distinct signals stipulate arterial or venous differentiation. Subsequent sprouting, known as angiogenesis , ensures expansion of the vascular network. Arteriogenesis then occurs, in which endothelial cell channels become covered by pericytes or vascular smooth muscle cells, which provide structure and regulate perfusion [2, 27].
Angiogenesis is a critical mechanism during embryonic development and under certain physiological circumstances in the adult, such as wound healing and formation of placenta during pregnancy [28, 29]. Angiogenesis is a complex process that is highly mediated by the endothelial cells that line the blood vessels [30].
In a fully developed (adult) mammal, when a quiescent vessel senses an angiogenic signal, pericytes detach from the vessel wall and set free from the basement membrane via proteolytic degradation mediated by matrix metalloproteases. Endothelial cells then loosen their junctions, and the nascent vessel dilates. VEGF increases the permeability of the endothelial cell layer, causing plasma proteins to extravasate from the vessel and to lay down a provisional extracellular matrix (ECM) scaffold. In response to integrin signalling, endothelial cells migrate onto this ECM surface. Proteases release angiogenic molecules stored in the ECM such as VEGF and FGF and also remodel the ECM. To build a perfused tube and prevent endothelial cells from moving all together in a deregulated fashion towards the angiogenic signal, one endothelial cell, named the tip cell, becomes selected to lead the tip in the presence of factors such as VEGF receptors, neuropilins and the NOTCH ligands, DLL4 and JAGGED1. Cells neighbouring the tip cell assume subsidiary positions as stalk cells, and divide to elongate the stalk [stimulated by NOTCH, NOTCH-regulated ankyrin repeat protein (NRARP), Wnt, PlGF and fibroblast growth factor (FGF)] and to establish the lumen of the blood vessel (mediated by VE-cadherin, CD34, sialomucins, VEGF and hedgehog) [31]. While tip cells have filopodia to sense environmental guidance cues such as ephrins and semaphorins, stalk cells release molecules such as EGF-like domain-containing protein 7 (EGFL7) into the ECM to convey spatial information about the position of their neighbours and to elongate the stalk [31]. Changes that occur in endothelial cell interactions with the ECM, as well as changes in cell-to-cell interactions are essential for the angiogenic process. Endothelial cells are linked to each other by tight and adherens-type junctions and are linked to the extracellular matrix by a variety of integrins and other adhesion molecules [32]. VEGF activates endothelial cells, in part through stimulating signal transduction pathways that regulate the enzymatic components of adhesion complexes. VEGF-induced tyrosine phosphorylation of VE-cadherins, a component of adherens-type cell-to-cell junctions, has been implicated as a key step in endothelial cell migration [33]. Experimental evidence supporting a role for VEGF in regulating cell-to-matrix interactions includes the findings that VEGF enhances the expression of integrins, and that neutralizing antibodies to v5 integrins block growth factor induced neovascularization [34, 35]. For a blood vessel to be perfectly functional, it must become mature and stable. Endothelial cells return to their quiescent state, and signals such as platelet-derived growth factor B (PDGF-B), angiopoietin 1 (ANG-1), transforming growth factor-β (TGF-β), ephrin-B2 and NOTCH induce the coverage of the newly formed blood vessel with pericytes and smooth muscle cells. Protease inhibitors known as tissue inhibitors of metalloproteases (TIMPs) and plasminogen activator inhibitor-1 (PAI-1) cause the deposition of a basement membrane and junctions are re-established to ensure optimal flow distribution. Under normal circumstances, vessels regress if they are unable to become perfused [31].
Normal angiogenesis is an extremely tightly regulated process involving not only a large number of stimulators, but also and very importantly inhibitors such as thrombospondin-1 (Tsp-1), angiostatin and endostatin [36–38]. Tsp-1 is a key negative regulator of angiogenesis inducing endothelial cell apoptosis , inhibiting migration and down regulating VEGF expression [39–43]. Angiostatin is a degradation product of plasminogen (Plg), constituted by kringles 1–3 of Plg. Angiostatin binds to proteins expressed on the surface of endothelial cells, such as annexin A2 heterotetramer (AIIt), angiomotin, integrin αvβ3, c-met and ATP synthase functioning as a negative regulator of these proteins and consequently inhibiting angiogenesis [44]. Endostatin is a 20-kDa C-terminal globular domain of collagen XVIII. A number of mechanisms have been proposed for endostatin anti-angiogenic activity, such as inhibition of phosphorylation of focal adhesion kinase (FAK) via binding to integrin α5β1, blockage of VEGF and Wnt signalling and binding and inactivation of metalloproteases [45].
4.7 Normal Versus Tumour Angiogenesis
Tumour angiogenesis is very different from normal angiogenesis in the sense that there is an excess of pro-angiogenic signalling that stimulates endothelial cell proliferation and migration, which is not accompanied by signals that lead to the recruitment and proliferation of perycites and smooth muscle cells. Also, in tumour angiogenesis the regulatory mechanisms that are responsible for “shutting down” neovascularisation in healthy tissues do not function normally. Angiogenesis inhibition in tumours is usually compromised since the transcription of the THBS1 gene that encodes for Tsp-1 is commonly impaired. THBS1 transcription is strongly induced by p53 [46]. Conversely, the loss of p53 function, observed in a large percentage of human tumours, leads to a substantial decrease in Tsp-1 protein expression within the tumour mass [47]. Oncogenes such as Myc, Ras, Src and Jun function in the opposite way inhibiting the transcription of the THBS1 gene [48–52]. Since constitutive activation of these oncogenes is frequently observed in tumours, this results in the inhibition of Tsp-1 protein expression and consequently also contributes substantially to the inhibition of anti-angiogenic mechanisms in cancer patients. As a consequence of the excessive pro-angiogenic signalling in conjunction with inhibition of anti-angiogenic mechanisms, tumour vasculature is marked by precocious capillary sprouting, convoluted and excessive vessel branching, distorted/ poorly structured and enlarged vessels, erratic blood flow, microhemorrhage, “leakiness” leading to accumulation of plasma in tissue areas close or inside the tumour, vessel collapse (which can create new hypoxic cores within the tumour) and abnormal levels of endothelial cell proliferation and apoptosis [53, 54].
4.8 The Role of Tumour Associated Cells in Angiogenesis
Presently it is widely recognized that tumour progression is not only the result of accumulating genetic alterations in cancer cells, and that the tumour microenvironment plays a key role in different aspects of tumourigenesis. The exacerbated pro-angiogenic signalling observed in tumours, particularly during hypoxia is not only due to signals coming from the cancer cells, but especially due to interactions between cancer cells, endothelial cells and tumour associated cells, such as macrophages and stromal cells which are crucial for tumour angiogenesis . Various angiogenic molecules produced by either cancer cells or tumour associated cells can directly bind to their cognate receptors on endothelial cells and thus initiate angiogenesis. Thus, a paracrine regulation of angiogenesis by secreted proteins is well-recognized.
For instance, VEGF secreted by the cancer cells will not only stimulate endothelial cell proliferation, but will also act as a chemoattractant for macrophages. Other growth factors including endothelin 2 secreted by endothelial cells and platelet-derived growth factor (PDGF), macrophage chemoattractant protein 1 (MCP-1) and colony-stimulating factor-1 (CSF-1) secreted by cancer cells and released from the ECM have also been reported to promote monocyte/macrophage recruitment to the tumour site [55, 56]. Macrophages constitute a major component of the tumour mass, where they are commonly termed tumour associated macrophages (TAMs). Macrophages shift their functional phenotypes in response to various microenvironmental signals generated by cancer and stromal cells. During tumour initiation, tumour-infiltrating macrophages usually show an M1 phenotype (IL-12high IL-10low), but at late-stage of tumour progression, TAMs generally switch to an M2 subset characterized by the IL-12low IL-10high phenotype [57]. Such TAMs (M2 subset) have been shown to provide a favourable microenvironment for tumour growth, survival and angiogenesis [58–60]. TAMs are recruited into hypoxic or necrotic areas of the tumour where they remove the tissue debris and stimulate repair processes [61, 62]. TAMs secrete a wide range of pro-angiogenic mediators, the most important of which being VEGF, but also including epidermal growth factor (EGF), basic fibroblast growth factor (bFGF), PDGF, thymidine phosphorylase (TP), angiopoietin receptor Tie2, angiogenic CXC chemokines (CXCL8/IL-8 and CXCL12, also known as stromal derived factor-1, SDF-1), angiogenesis-associated factors such as transforming growth factor beta (TGF-β), tumour necrosis factor alpha (TNF-α) and adrenomedullin (ADM), further promoting tumour angiogenesis [59, 63–66]. TAMs also secrete proteolytic enzymes such as plasmin, urokinase-type plasminogen activator (uPA) (activator of the protease plasmin), and metalloproteases, MMP-1, MMP-2, MMP-3, MMP-7, MMP-9 and MMP-12, whose combined action induces degradation of the basement membrane and ECM components, release of sequestered growth factors from the ECM, destabilization of the vasculature as well as migration and proliferation of endothelial cells contributing significantly in this way to tumour angiogenesis [59, 66–69].
Neutrophils inflammatory cells have also been shown to infiltrate tumours and promote angiogenesis [70]. CXCL8, which is abundantly produced by tumour cells, represents a potent chemoattractant for the recruitment of neutrophils to the tumour mass. CXCL8 is also associated with angiogenesis by directly activating the CXCR2 receptor on endothelial cells [71]. Activated neutrophils secrete VEGF, metalloproteases that degrade and remodel the ECM (e.g. MMP9) and chemokines, CXCL8 and CXCL1 contributing to tumour angiogenesis [72–74].
Natural killer (NK) cells are also recruited to the tumour site. The tumour microenvironment is able to affect NK functionality by a wide array of cytokines and soluble factors (e.g. TGF-β, prostraglandin E2 (PGE2), VEGF), that either inhibit their cytotoxic function or promote a pro-tumourigenic and pro-angiogenic phenotype [75, 76]. Recent reports have shown that tumour infiltrating NK cells produce elevated levels of VEGF, PlGF, IL-8 and induce endothelial cells chemotaxis and tube formation [76].
The recruitment and activation of mast cells (MCs, also known as mastocytes) to the tumour site has been shown to be mainly mediated by tumour-derived stem cell factor (SCF) and its receptor c-kit on MCs [77]. Mast cells contribute to the angiogenic switch in tumours through the production of diverse pro-angiogenic growth factors, cytokines and chemokines, including VEGF, angiopoietin-1, FGF-2, IL-8 and TGF-β [78, 79]. Proteases produced by mast cells, such as tryptase, chymase, cathepsin G, elastase and collagenase, promote angiogenesis and are currently becoming targets for anti-angiogenic therapy [78, 80–83].
The fibroblasts within the tumour mass, also known as cancer-associated fibroblasts (CAFs) also contribute significantly to tumour angiogenesis . CAFs are of multiple origins: they can originate from resident fibroblasts, mesenchymal stem cells or mutated fibroblasts [84]. CAFs are able to produce cytokines and chemokines favouring inflamatory cells infiltration and consequently promoting angiogenesis and metastasis. SDF-1 producing CAFs play a key role in the recruitment of endothelial cells to the tumour site [85, 86]. CAFs are also able to produce CXCL14, this in turn enhances interactions with tumour cells and favour macrophage infiltration and M2 subset polarization [87]. Recent studies have shown that CAFs associated to incipient neoplasia exhibit a pro-inflammatory signature, characterized by an over-expression of SDF-1, IL-6 and IL-1β that lead to the recruitment of pro-angiogenic macrophages and sustain tumour growth [87]. In addition, CAFs also secrete FGF which is a well characterized pro-angiogenic growth factor [88].
4.9 Anti-angiogenic Cancer Therapy
It is currently accepted that the main pro-angiogenic factor secreted within the tumour mass is VEGF. For this reason several anti-angiogenic drugs have been developed to target VEGF or its receptor, VEGFR-2. A variety of drugs, such as antibodies against VEGF or its receptor, engineered proteins that mimic VEGFRs and small molecule receptor tyrosine kinase inhibitors that preferentially target VEGFR-2 (VEGFR-2/flk-1/KDR) with high affinity effectively prevent the growth of many mouse tumours and tumour xenografts [31, 89–93]. Unfortunately, however, the striking benefits of anti-VEGF/VEGFR therapy observed when treating mouse tumours have not been translated to the clinic. These drugs have had only modest effects on human cancers.
4.10 Anti-angiogenic Chemotherapeutics
Currently there are several Food and Drug Administration (FDA) approved anti-angiogenic chemotherapeutic drugs, including bevacizumab (Avastin; Genentech), aflibercept, axitinib, imatinib, pazopanib, regorafenib, sorafenib, sunitinib, and vandetanib. The best characterized and most widely used anti-angiogenic chemotherapeutic agent is bevacizumab, a humanized antibody against VEGF. Like bevacizumab, aflibercept is an inhibitor of VEGF. Aflibercept is a recombinant fusion protein consisting of VEGF-binding domains for the extracellular moiety of human VEGF receptors 1 and 2 that are fused to the Fc portion of the human IgG1 immunoglobulin; acting as a decoy VEGFR (VEGF trap) [94]. Axitinib, imatinib, pazopanib, regorafenib, sorafenib, sunitinib and vandetanib are multi-targeted receptor tyrosine kinase inhibitors that inhibit pro-angiogenic receptors, such as VEGFRs, FGFRs and PDGFRs [94]. Although these anti-angiogenic chemotherapeutics either alone or in combination with other drugs have been shown to improve progression-free survival and overall survival in cancer patients, their efficacy is still distant from what was anticipated and is usually accompanied with serious side effects. In addition, variable results have been observed in the treatment of different types of cancers with these drugs, suggesting that the sensitivity and efficacy of anti-angiogenic therapy might be cancer specific [95].
4.11 Potential Pitfalls of Anti-angiogenic Therapy
A number of explanations have been put forward in order to explain the modest effectiveness of anti-VEGF/VEGFR therapy in cancer patients compared to laboratory mice. An obvious explanation is that cancer patients are often elderly and very ill, in contrast with the young, relatively healthy tumour-bearing laboratory mice. Furthermore, mice usually take much higher chemotherapeutic dosages compared to cancer patients, without taking into account toxic side effects. Another likely reason for the limited effectiveness of anti-VEGF/VEGR therapy is that it does not result in the killing of all tumour cells; as such the remaining cancer cells rendered hypoxic by a compromised blood supply are stimulated to produce and secrete increased amounts of VEGF that may overwhelm anti-VEGF/VEGFR therapy, especially when accompanied by increased expression of matrix components that bind and sequester VEGF, protecting it from anti-VEGF drugs [96]. Hypoxic cancer cells also produce a plethora of other growth factors and cytokines, which have the capacity to stimulate new blood vessel formation and growth, including FGF, PDGF, HGF, EGF, IL-8, IL-6, Ang-2, SDF-1, PDGF-C, CXCL6, and others, as well as their receptors. The recruitment of vascular progenitor cells and pro-angiogenic immune cells (e.g. macrophages, mastocytes, NK cells, neutrophils) that can serve as a rich source of growth factors, cytokines and chemokines constitutes another possible mechanism for the lack of success observed with anti-VEGF/VEGFR cancer therapy [97, 98]. Several studies have also shown that VEGFR inhibitors are actually highly effective in preventing the development of the spontaneous Rip-Tag tumour and in inhibiting its early growth, but are much less beneficial in regressing tumours with an already established vasculature [97, 99]. Thus, in mice as in patients, anti-VEGF/VEGFR therapy was found to be less effective in advanced disease. Bergers and Hanahan attributed the failure of late therapy to the maturing of the vasculature with increased pericyte coverage and found that addition of a receptor tyrosine kinase inhibitor that targeted PDGFR-β (highly expressed on pericytes) improved anti-VEGFR therapy [97]. Many other reports indicate that immature vessels are preferentially susceptible to anti-VEGF/VEGFR therapy [97, 99, 100]. There is microvascular heterogeneity within tumours, and not all activated endothelial cells express the same cell surface markers. Therefore, the pharmaceutical targeting of a specific marker may not effectively inhibit tumour progression.
It is becoming increasingly clear that in order to develop highly efficient anti-angiogenic therapies, we probably need to target several pro-angiogenic key molecules simultaneously to effectively hinder tumour vascularization. Also, combinational therapies involving anti-angiogenic drugs directed at inhibiting vessel formation in conjunction with chemotherapeutics that specifically target/kill cancer cells have shown promising results [94, 95].
Once tumour angiogenesis is established the high density of blood vessels within the tumour site provides not only oxygen and nutrients that allow the tumour to grow, but also an escape route for the cancer cells (metastasis), for these reasons tumour angiogenesis is closely linked to poorer clinical outcome for cancer patients [2].
Angiogenesis constitutes the first/initial step of the tumour invasion/metastatic cascade, simultaneously with local invasion of connective tissue (to which endothelial cells contribute significantly, especially at the initial stages of tumour development); the next step of the invasion/metastatic cascade is intravasion, where cancer cells enter the blood vessels; followed by transport of the cancer cells in the blood stream; extravasion is then complied by the adhesion of cancer cells to the blood vessel and entry into tissues/organs in a distinct location from the primary tumour; formation of micrometastasis follows, which is the establishment of the cancer cells in these new tissues/organs and finally colonization comprises the proliferation of the newly established cancer cells in order to form large masses, macrometastasis.
References
1.
2.
3.
Bertout JA, Patel SA, Simon MC (2008) The impact of O2 availability on human cancer. Nat Rev Cancer 8:967–975CrossRefPubMedCentralPubMed
4.
Sadri N, Zhang PJ (2013) Hypoxia-inducible factors: mediators of cancer progression; prognostic and therapeutic targets in soft tissue sarcomas. Cancers (Basel) 5:320–333
5.
Related posts:
 Anti-cancer Drugs: Discovery, Development and Therapy
Anti-cancer Drugs: Discovery, Development and Therapy
 Diagnosis and Management of Gestational Trophoblastic Neoplasia
Neoplastic Epidural Spinal Compression Cord Compression
An Overview of Treatment for Cervical Cancer with Emphasis on Immune Cell-Based Therapies
Diagnosis and Management of Gestational Trophoblastic Neoplasia
Neoplastic Epidural Spinal Compression Cord Compression
An Overview of Treatment for Cervical Cancer with Emphasis on Immune Cell-Based Therapies
 Chemotherapy-Induced Nausea and Vomiting: Molecular Mechanisms and Clinical Approaches
Chemotherapy-Induced Nausea and Vomiting: Molecular Mechanisms and Clinical Approaches
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


