Outline
Embryology, 460
Esophagus and Stomach, 461
Esophageal Atresia, 461
Small Intestine, 465
Duodenal Obstruction, 465
Echogenic Bowel, 465
Jejunoileal Obstruction, 469
Meconium Peritonitis, 470
Rectum, 471
Anorectal Malformations, 471
Liver, Spleen, and Gallbladder, 475
Cysts, 479
Differential Diagnosis, 479
Ovarian Cysts, 480
Enteric Duplication Cysts, 481
Rare Causes of Cysts, 481
Ventral Wall Defects, 483
Omphalocele, 483
Gastroschisis, 493
Bladder and Cloacal Exstrophy/OEIS Complex, 496
Pentalogy of Cantrell, 497
Limb–Body Wall Complex/Amniotic Band Syndrome, 497
Urachal Cyst, 497
Summary of Key Points
- •
Detection of gastrointestinal tract obstruction is often difficult. Duodenal and small intestinal obstructions are easily visualized, but usually not until the third trimester, whereas esophageal atresia and anal atresia may be difficult to detect at any gestational age.
- •
Echogenic small intestine is a nonspecific finding that is associated with an increased risk for Down syndrome, cystic fibrosis, congenital cytomegalovirus (CMV) infection, fetal growth restriction (FGR), and fetal demise. However, unless there is an increased a priori risk, most fetuses with isolated echogenic bowel have a normal outcome.
- •
Several sonographic findings in the fetal abdomen can be associated with cystic fibrosis, including echogenic bowel, meconium peritonitis, meconium ileus, and absent gallbladder. If detected, these findings should be correlated with genetic screening results for cystic fibrosis, which is routinely offered in pregnancy.
- •
Ventral wall defects can often be diagnosed between 11 and 14 weeks, but be aware that isolated small omphaloceles may spontaneously resolve and can perhaps best be thought of as delayed resolution of physiologic bowel herniation.
- •
Omphalocele can almost always be distinguished from gastroschisis because it involves the umbilicus, is covered by a membrane, and often contains liver. Gastroschisis involves free loops of externalized bowel that have herniated to the right of the umbilical cord insertion site.
- •
Omphalocele is more likely than gastroschisis to be associated with aneuploidy and other anomalies. Although gastroschisis is typically isolated, affected fetuses have an increased risk of FGR and fetal demise and may have a prolonged course in the neonatal nursery.
- •
The risk of aneuploidy in a fetus with an omphalocele is directly related to the presence of associated anomalies, and inversely related to the gestational age.
Embryology
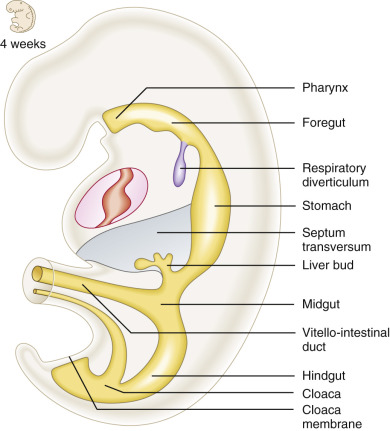
The gastrointestinal tract begins its development early in the 3rd and 4th weeks, as longitudinal and lateral folding of the embryo results in incorporation of the dorsal part of the yolk sac. As this occurs, the endoderm germ cell layer is incorporated into the embryo and forms the primordial (primitive) gut tube ( Fig. 14-1 ). This primitive gut is closed at the cranial end by the oropharyngeal membrane and at the caudal end by the cloacal membrane. The endoderm of the primitive gut gives rise to most of the epithelium and glands of the digestive tract. The epithelium at the cranial and caudal ends of the tract is derived from the ectoderm of the stomodeum (primitive mouth) and proctodeum (anal pit), respectively. The muscular layer, connective tissue, and other layers of the wall of the digestive tract are derived from the splanchnic mesenchyme surrounding the primitive gut. In the anterior portion of the embryo, incorporation of the endoderm into the head fold results in the formation of the foregut, whereas in the posterior portion of the embryo the hindgut forms. The foregut is divided into a cranial and a caudal portion. The cranial portion develops in the head and neck as the pharynx. In the middle region, the midgut forms, and initially this is in direct continuity with the yolk sac. In addition to the primitive gut tube, the endodermal layer also gives rise to the parenchyma of the two large glandular organs associated with the gastrointestinal duct, the liver and pancreas.
Esophagus and Stomach
The normal esophagus is a collapsed structure that is not routinely imaged. Although it is possible to see the esophagus with a high-frequency transducer after 19 weeks in the majority of cases, this is not part of routine sonography. The presence of a patent and normally functioning esophagus is usually inferred by noting fluid in the fetal stomach, the so-called “stomach bubble.” This normally appears as a sonolucent area in the left upper quadrant of the abdomen ( Fig. 14-2A and C ), which can be seen as early as the first trimester. There may be echogenic material in the fetal stomach bubble, thought to be debris in the amniotic fluid that has been swallowed by the fetus and is of no clinical significance.
The stomach is located on the right side of the abdomen in situs inversus totalis , which involves left-right reversal of otherwise normal gross anatomy such that the heart, stomach, and spleen are to the right and the liver is to the left. In about one fourth of cases situs inversus totalis is associated with primary ciliary dyskinesia (Kartagener syndrome), which can cause respiratory distress in newborns, chronic respiratory tract infections, and male infertility.
The stomach can be on the left, in the middle, or on the right side of the abdomen in heterotaxy syndromes , a group of disorders of laterality that include polysplenia (bilateral left-sidedness) and asplenia (bilateral right-sidedness). Most cases of heterotaxy syndrome are associated with congenital heart disease.
The stomach is frequently displaced by diaphragmatic hernia. It is usually in the thorax, but in milder cases can remain in the abdomen.
The amount of fluid in the stomach is highly variable among normal fetuses as, over time, in an individual fetus the stomach cyclically fills after fetal swallowing and empties through the pyloric valve into the duodenum. A larger than average quantity of fluid in the stomach is common and is generally of no clinical significance as long as there is a normal amount of amniotic fluid and the stomach bubble does not cross the midline, which would suggest duodenal obstruction. Pyloric atresia is associated with a large stomach bubble, but is extremely rare and is usually accompanied by polyhydramnios and possibly esophageal dilation. Pyloric stenosis does not become symptomatic until after the child is born and generally cannot be detected in utero.
A small or absent stomach bubble in the absence of polyhydramnios is most often a transient finding seen in a normal fetus. Having the patient return in an hour or so, if at all possible, should cause the patient less anxiety and inconvenience than having her return for a follow-up scan in 1 or 2 weeks. A persistently small or absent stomach bubble, particularly when accompanied by polyhydramnios, suggests an abnormality such as esophageal atresia.
Esophageal Atresia
Esophageal atresia is an interruption of the esophagus such that the upper esophagus ends in a blind pouch, most often at or above the tracheal bifurcation ( Fig. 14-3 ). The prevalence of esophageal atresia is about 2.4 per 10,000. In as many as 90% of cases, esophageal atresia is associated with a tracheoesophageal fistula, which is almost always from the trachea to the distal esophagus.
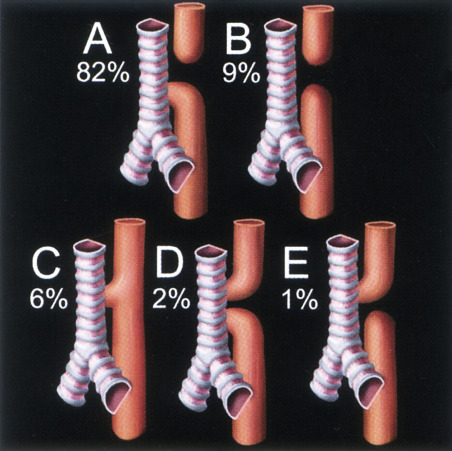
Associated abnormalities are common. In the EUROCAT (European Registry of Congenital Anomalies and Twins) series of 1222 cases over a 20-year period, esophageal atresia was isolated in less than half of cases ( Table 14-1 ). Associated abnormalities are even more common in prenatal compared to pediatric series of esophageal atresia and indeed may dominate the sonographic picture and result in a poorer prognosis. In an early series, 7 of 16 cases of correctly diagnosed esophageal atresia had trisomy 18, and only 4 survived the neonatal period. Two more recent series found additional abnormalities in 76% of prenatally diagnosed cases.
| Cause | Incidence |
|---|---|
| Aneuploidy | |
| Trisomy 18 | 6% |
| Down syndrome | 2% |
| Other | 1% |
| Genetic Syndromes | |
| CHARGE * | 1% |
| Other | 5% |
| VACTERL * | 10% |
| Unclassified multiple malformations | 31% |
| Isolated | 45% |
* See Table 14-3 .
Sonographic Diagnosis
Esophageal atresia is suspected when the amount of fluid in the stomach is less than normal, particularly in the presence of polyhydramnios. The stomach bubble may not be completely absent, because when there is a distal tracheoesophageal fistula (present in the majority of cases) tracheal fluid can flow across the fistula into the distal esophagus and stomach. Even in the setting of esophageal atresia without fistula, gastric secretions may produce a small amount of fluid in the stomach (see Fig. 14-2 ). Unfortunately, there are no good criteria that define the lower limit of normal size for the fetal stomach. Retrospective reviews describe a “small or absent stomach bubble” or an “absent or collapsed fetal stomach” without quantification.
Assessment of stomach size can be subjective and presents a tradeoff between sensitivity and specificity. Sonologists who consider very small stomach bubbles to be normal will occasionally miss an anomalous fetus. Conversely, sonologists who raise concern for these same small stomach bubbles will have many more false positive diagnoses, resulting in more anxious patients, an increased number of follow-up sonograms, and potentially unnecessary amniocenteses.
A completely absent stomach bubble is more likely than a small stomach bubble to represent esophageal atresia, particularly in the presence of polyhydramnios. For example, in one study esophageal atresia was present in all five fetuses with polyhydramnios and an absent stomach bubble but was found in only three of six fetuses with polyhydramnios and a small stomach bubble. As expected, a small stomach bubble with normal amniotic fluid volume was often a transient finding in a normal fetus.
Care must be taken to confirm that the stomach is not in an unusual location, as may be the case with situs inversus, heterotaxy syndrome, diaphragmatic hernia, or ventral wall defect. A persistently absent or very tiny stomach bubble suggests an inability of the fetus to swallow amniotic fluid and transport it to the stomach. There are several possible causes in addition to esophageal atresia ( Table 14-2 ).
| Oligohydramnios from any cause (e.g., preterm premature rupture of membranes, bilateral renal disease, urethral obstruction, donor twin in twin-twin transfusion syndrome) presumably because there is less fluid to swallow. |
| Neuromuscular disorder that impairs active swallowing: any condition that is associated with hypokinesia and/or arthrogryposis. |
| Abnormalities of the face or neck that can interfere with active swallowing or cause a mechanical obstruction: micrognathia (often associated with hypokinesia syndromes), orofacial cleft, epignathus (teratoma originating from the mouth), neck mass (teratoma, large goiter). |
| Esophageal atresia with or without tracheoesophageal fistula. |
Therefore, sonographic evaluation of the fetus with a small or absent stomach bubble should always involve meticulous inspection of the rest of the anatomy with particular attention to amniotic fluid volume, fetal activity, and the fetal face, neck, and chest. A 2010 series reported a wide variety of conditions in association with persistent nonvisualization of the fetal stomach: esophageal atresia was found in only 8/48 cases (17%) after excluding those with oligohydramnios or aberrant location of the stomach. Seven fetuses were ultimately found to be normal. Fetal akinesia sequence was present in six cases.
The detection rate of esophageal atresia is poor. There were 152 cases of esophageal atresia with complete data in Northern England between 1985 and 1997; only 14 were prenatally diagnosed, for a detection rate of 9%. The EUROCAT registry reported that the detection rate increased only slightly over time, from 26% during the years 1987 to 1996 to 36% in the following decade. The best results were obtained in Norway: 46% of 46 cases were detected, although 32 of these cases were referred because of sonographic abnormalities. A recent, more representative review of children treated for esophageal atresia in Texas between 2002 and 2014 showed that only 16% of 91 cases were prenatally diagnosed, suggesting that the “real world” detection rate remains low.
In addition, prenatal diagnosis of esophageal atresia, when made, tends to occur late in gestation. Approximately one third are diagnosed prior to 28 to 30 weeks, with median gestational age of in utero diagnosis of esophageal atresia of 31 to 32 weeks (see Fig. 14-2 ).
As previously noted, the sonographic diagnosis of esophageal atresia is often incorrect. Esophageal atresia was found in 17% of fetuses with a small or absent stomach bubble; this increased to 34% to 44% when there was also increased amniotic fluid volume. In a more recent study, esophageal atresia was found in 8/18 euploid fetuses that had persistent nonvisualization of the stomach and polyhydramnios; 2 fetuses were normal, and the remaining 8 had a variety of other problems. In a series of 22 fetuses with suspected esophageal atresia, the diagnosis was more likely to be correct if subsequent scans showed a persistently absent stomach bubble and development of polyhydramnios ; 11 fetuses in this study did not have esophageal atresia, and 5 were found to be normal.
In an effort to improve the accuracy of prenatal sonographic diagnosis of esophageal atresia, several authors have suggested use of the pouch sign representing the proximally dilated esophagus in the neck or thorax. This sign was present in 43% of fetuses with esophageal atresia, and most authors feel that it is very specific, so when seen, the diagnosis is most frequently correct. However, an esophageal pouch should not be confused with a normal pharynx; false positive results have been reported.
More recent studies have utilized magnetic resonance imaging (MRI); at a mean gestational age of 30 weeks an esophageal pouch was seen in 10/12 fetuses (83%) with and in 0/11 fetuses without postnatally confirmed esophageal atresia. In another recent (2014) study at a mean gestational age of 32 weeks, an abnormal MRI was correct in all 8 cases, whereas a normal MRI was correct in only 5 of 7. Note that both of these series evaluated fetuses in the third trimester.
Antepartum Management
When esophageal atresia is suspected because of a persistently small or absent stomach bubble, a careful ultrasound examination should be performed, with special attention to the following:
- 1.
Fetal activity to exclude the possibility of diminished fetal swallowing due to fetal hypokinesia
- 2.
The face and neck to exclude the possibility of an anatomic obstruction
- 3.
Findings associated with VACTERL (vertebral anomalies, anal atresia, cardiac defects, tracheoesophageal fistula and/or esophageal atresia, renal anomalies, and limb defects) ( Table 14-3 ): including vertebral bodies, rectum and anus, heart, kidneys, extremities (especially radii and thumbs) and umbilical arteries
TABLE 14-3
Characteristics of VACTERL Association and CHARGE Syndrome
VACTERL is an acronym that refers to an association of nonrandom co-occurring structural anomalies involving multiple body parts and organ systems. a
V: vertebral anomalies such as segmentation defects, e.g., hemivertebrae (60%-80%)
A: anorectal malformations, e.g., anal atresia (55%-90%)
C: cardiac abnormalities (40%-80%)
TE: esophageal atresia with or without tracheoesophageal fistula (50%-80%)
R: renal abnormalities (50%-80%)
L: limb malformations, such as radial ray defects (40%-50%)
By common agreement, at least three anomalies must be present to warrant this diagnosis/label, without evidence of overlapping genetic conditions, such as Fanconi anemia.
Additional anomalies are frequently associated with VACTERL. For example, single umbilical artery is seen in 20% of cases. b
The prevalence of VACTERL association is reported to be 1 : 10,000 to 1 : 40,000. a The underlying cause is not known, although changes in genes coding for transcription factors have been implicated. c Although most cases are sporadic, 9% of probands have a first-degree relative with at least one component of this association. d
CHARGE syndrome is caused by a mutation in CHD7 gene.
C oloboma of the eye
H eart defects
A tresia choanae (also known as choanal atresia)
R etardation of growth and/or development
G enital and/or urinary abnormalities
E ar abnormalities
Although not part of the acronym, tracheoesophageal fistula is found in 20% of cases (see Fig. 14-2 ). e
a Solomon BD: VACTERL/VATER association. Orphanet J Rare Dis 6:56, 2011.
b de Jong EM, Felix JF, Deurloo JA, et al: Non-VACTERL-type anomalies are frequent in patients with esophageal atresia/tracheo-esophageal fistula and full or partial VACTERL association. Birth Defects Res A Clin Mol Teratol 82(2):92-97, 2008.
c Shaw-Smith C: Genetic factors in esophageal atresia, tracheo-esophageal fistula and the VACTERL association: roles for FOXF1 and the 16q24.1 FOX transcription factor gene cluster, and review of the literature. Eur J Med Genet 53:6-13, 2010.
d Solomon BD, Pineda-Alvarez DE, Raam MS, Cummings DA: Evidence for inheritance in patients with VACTERL association. Hum Genet 127(6):731-733, 2010.
e Zentner GE, Layman WS, Martin DM, Scacheri PC: Molecular and phenotypic aspects of CHD7 mutation in CHARGE syndrome. Am J Med Genet A 152A:674-686, 2010.
- 4.
Findings associated with aneuploidy (especially Down syndrome and trisomy 18)
Because of the high prevalence of associated congenital heart disease, a fetal echocardiogram is recommended. An MRI can be considered, both to evaluate the esophagus and to search for additional anomalies not detected by ultrasound examination. The possibility of fetal aneuploidy should be addressed. If amniocentesis is performed, testing for the CHD7 gene associated with CHARGE (see Table 14-3 ). syndrome can be considered.
Serial sonograms should be obtained to search for additional anomalies and evaluate fetal growth. The diagnosis of esophageal atresia typically becomes more evident over time, with a persistently absent stomach bubble and development of polyhydramnios (see Fig. 14-2 ). A pediatric surgery consult is advised, and delivery should be at a tertiary care center.
Neonatal Management
Feeding should be withheld. Passage of a catheter into the stomach is attempted and a radiograph is obtained. In the presence of esophageal atresia, the tip of the catheter is kinked at about T2 to T4.
Repair of the most common form of esophageal atresia, with a distal tracheoesophageal fistula, can be done via either thoracotomy or thorascopic approach. If there is no distal fistula, repair can be more complicated and may require initial placement of a gastrostomy.
Survival rate for full-term infants without associated anomalies approaches 100%. Complications can include anastomotic leaks, esophageal stricture, and recurrent tracheoesophageal fistula. Other long-term problems include tracheomalacia, disordered esophageal peristalsis, gastroesophageal reflux, vocal cord dysfunction, and respiratory problems.
Small Intestine
Duodenal Obstruction
Duodenal obstruction can be intrinsic, most commonly because of atresia, or extrinsic, from constriction. An annular pancreas may occur in conjunction with an intrinsic defect rather than representing a true constricting lesion. A duodenal web can cause partial obstruction. The prevalence of duodenal obstruction is about 1.8 per 10,000 births.
Ultrasound Diagnosis
The characteristic “double bubble sign” of duodenal obstruction, usually seen in conjunction with polyhydramnios, was one of the first fetal anomalies that could be detected by ultrasound. However, a more specific diagnosis requires demonstration of continuity of the dilated duodenum with fluid in the stomach, crossing the midline of the fetus ( Fig. 14-4 ). If such continuity cannot be established, other causes of an upper abdominal cyst (such as choledochal, mesenteric, hepatic, or enteric duplication cyst) should be considered. The differential diagnosis also includes duodenal duplication.
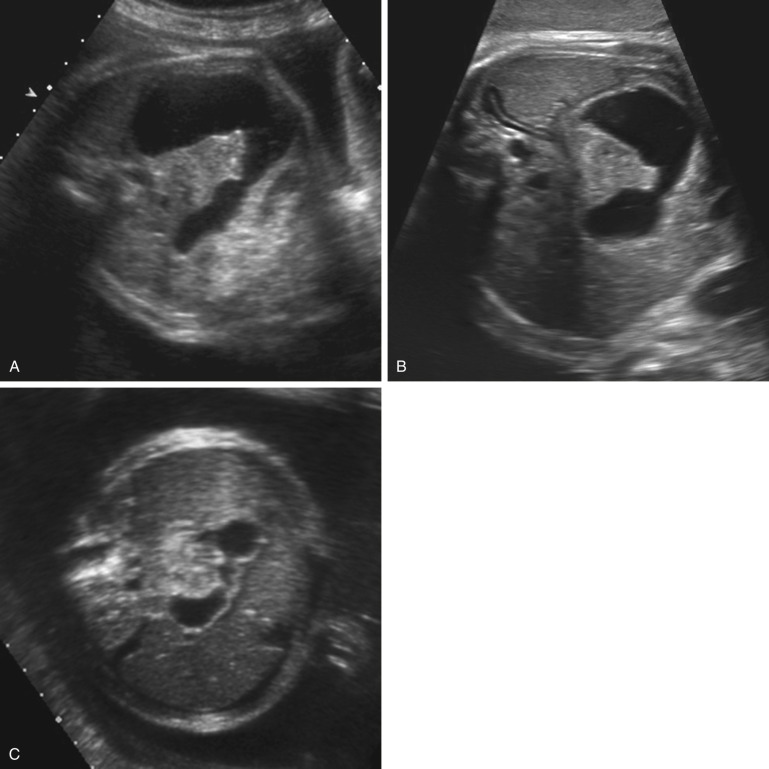
Cases of transient duodenal dilation have been reported, presumably caused by transit of fluid across the pyloric valve, so assessment of suspected dilation should include documentation for a reasonable length of time.
Duodenal obstruction can rarely, if ever, be diagnosed in the first trimester. At the routine 20-week obstetric sonogram, the detection rate may be as high as 50%, but like other obstructions of the gastrointestinal tract, this diagnosis is more reliably made in the third trimester.
Associated Anomalies
Up to one half of children with duodenal obstruction have Down syndrome. This is not surprising considering that the relative risks of duodenal atresia and annular pancreas for Down syndrome are 264 and 430, respectively. That is, the finding of duodenal obstruction increases the odds that a fetus has Down syndrome by a factor of at least 264.
In addition to Down syndrome, duodenal obstruction is associated with a wide variety of structural abnormalities, in particular congenital heart disease, additional intestinal problems such as malrotation and atresia, and all of the anomalies that are part of the VACTERL association (see Table 14-3 ).
Prognosis
As is true with many conditions, the prognosis for duodenal obstruction is highly dependent on the presence or absence of associated anomalies. In isolated duodenal obstruction, survival rate approaches 100% ; there is a wide variation in mortality rate in complicated cases, dependent on the nature of the associated anomalies.
Antenatal Management
At the time of diagnosis, a detailed search for additional abnormalities should include a fetal echocardiogram. The risk of aneuplopidy should be addressed. Duodenal obstruction is associated with a high rate of prematurity, possibly because of polyhydramnios, and an increased rate of unexpected fetal demise. Antenatal surveillance is sometimes considered, although the benefit is unclear. A pediatric surgery consult is advised, and delivery should be at a tertiary care center.
Neonatal Management
Fluid resuscitation and gastric decompression are followed by surgical repair, which can be either laparoscopic or by open duodeno-duodenostomy. Long-term survival is excellent for term infants without associated anomalies but complications such as gastroesophageal reflux and delayed gastric emptying can occur.
Echogenic Bowel
Definition
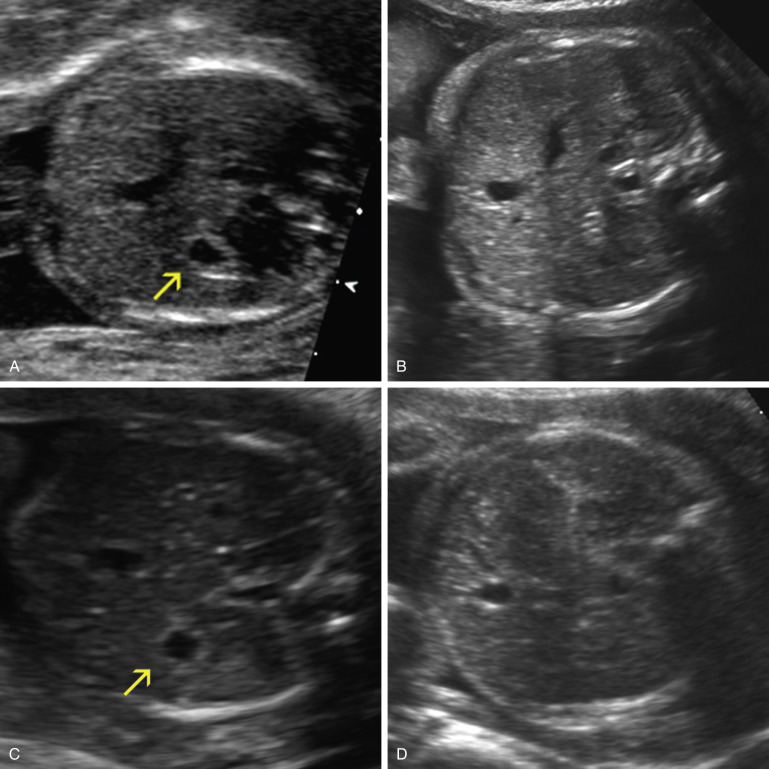
On second trimester ultrasound imaging, the fetal liver, lung, small intestine, and bone demonstrate increasing echogenicity in that order ( Fig. 14-5A ). In the 1990s it was noted that sometimes the echogenicity of small intestine is increased, such that it appears as bright as bone. This was referred to as “echogenic bowel,” a term often used interchangeably with echogenic small intestine. At that time, standard transducers used for obstetric ultrasound imaging had frequencies of 3.5 to 5 MHz and were without harmonic signal processing. Present-day transducer frequencies are typically 5 to 8 MHz and harmonics are routinely employed. These newer transducers offer improved resolution and thereby enhance prenatal diagnosis. They also make the disparity in echogenicity between small intestine and liver more conspicuous, thereby making the diagnosis of echogenic bowel more difficult ( Fig. 14-5B and C ).
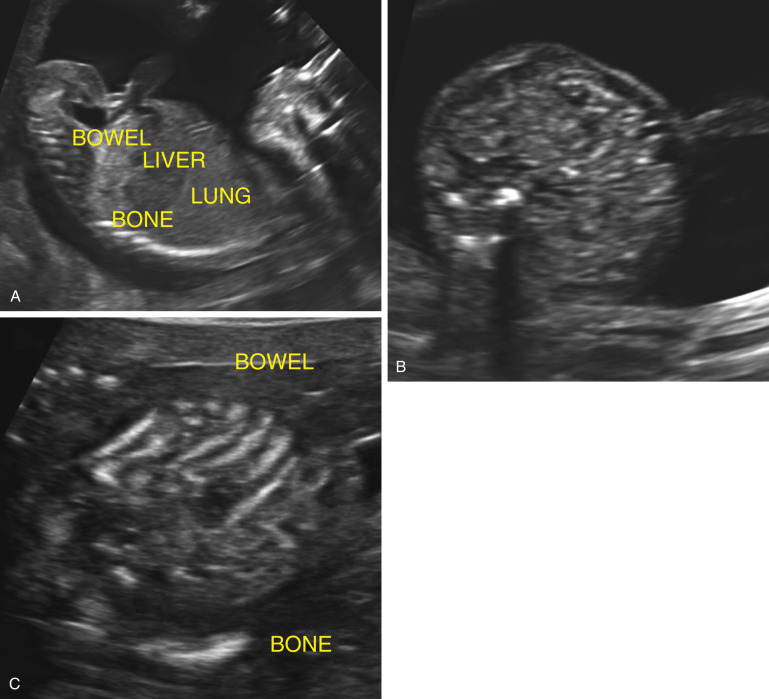
For these reasons, it is now recommended that when echogenic small intestine is suspected, a low-frequency transducer (≤5 MHz) should be used. Some authors suggest that the gain be turned down to allow for comparison between the echogenicity of small intestine and bone. Despite these caveats, the detection of echogenic small intestine remains very subjective, more than most other sonographic observations ( Fig. 14-6A ). There are no standardized criteria for the sonographic diagnosis of echogenic small intestine.
A 2011 review states that the prevalence of echogenic small intestine in routine second trimester sonograms ranges from 0.2% to 1.8%. This ninefold range in prevalence attests to the subjectivity of this finding.
Etiology
Echogenic small intestine is thought to be due to abnormal characteristics of intraluminal bowel contents or edema of the bowel wall. Factors such as decreased amniotic fluid volume, presence of meconium, intestinal hypomotility because of ischemia, and swallowed blood after intra-amniotic bleeding have been associated with echogenic bowel.
Association With Adverse Outcomes ( Table 14-4 )
Down Syndrome.
The odds that a fetus has Down syndrome is increased in the presence of echogenic small intestine. The amount that it is increased is defined by the likelihood ratio (LR). Owing in part to the subjective nature of echogenic bowel, LR values as disparate as 1.7 and 5.5 to 6.7 have been cited in a meta-analysis and in the executive summary of a National Institutes of Health (NIH)-sponsored workshop, respectively. In addition, the positive LR of an isolated finding (such as echogenic bowel) depends on the product of the negative LRs of all of the other sonographic markers, referred to as “soft signs.” The earlier studies cited by the NIH workshop did not include evaluation of the nasal bone, which has a negative LR of approximately 0.5. Conversely the meta-analysis considered evaluation for an aberrant subclavian artery, which is not frequently done. If evaluation of the nasal bone is added to the NIH workshop estimate, the LR for isolated echogenic bowel decreases to about 3, and if evaluation of the subclavian artery is removed from the meta-analysis, the LR for isolated echogenic bowel increases to 2.4. Thus the two estimates can be more closely reconciled.
| Aneuploidy, in particular Down syndrome |
| Cystic fibrosis |
| Growth restriction and fetal demise |
| Congenital infection, in particular cytomegalovirus (CMV) |
| Gastrointestinal obstruction |
Cystic Fibrosis.
Cystic fibrosis has been detected in about 8% of fetuses with echogenic small intestine. However, these patients were not previously screened for cystic fibrosis carrier status, which can detect approximately 90% of heterozygotes in the caucasian population; offering such screening is the standard of care in the United States. The frequency of cystic fibrosis in a prescreened population with echogenic bowel would clearly be much less. In some cases, sequencing of the cystic fibrosis transmembrane receptor gene may be helpful in patients with echogenic fetal bowel and normal cystic fibrosis screening results.
Congenital Infection.
In one study, there were reported to be sonographic findings in 30 of 69 cases of congenital CMV infection, including 9 cases of echogenic small intestine. Other sonographic findings associated with congenital CMV infection included growth restriction, head circumference below the 5th percentile, brain calcifications, and ventriculomegaly. An earlier study demonstrated similar results: 7 of 154 fetuses (5%) with documented congenital CMV infection had echogenic bowel, whereas 131 fetuses (85%) had no sonographic findings. Conversely, the likelihood of CMV infection in a fetus with echogenic small intestine is at most 3% and the association with other infections is even less.
Fetal Growth Restriction and Stillbirth.
In recent years, increasing attention has been directed toward the association between echogenic small intestine and obstetric complications. The likelihood of fetal demise and of FGR was increased by a factor of 9.6 and 2.1, respectively, in fetuses with echogenic small intestine. The median gestational age at fetal demise was 24 weeks, and in this study all spontaneous fetal losses that were observed in the setting of isolated echogenic small intestine occurred at or before 30 weeks. A subsequent study reported very similar findings, with an increase in the likelihood of growth restriction from 1.3% to 9.9% and an increase in the likelihood of fetal demise from 0.5% to 8.9% in fetuses with isolated echogenic small intestine. Again, the median gestational age at fetal demise was 24 weeks and all spontaneous losses occurred prior to 30 weeks. These data suggest that the advantage of antenatal surveillance starting at 32 weeks may be limited.
Additional factors that may further increase the likelihood of obstetric complications in fetuses with echogenic small intestine include elevated maternal serum alpha-fetoprotein and abnormal uterine artery Doppler.
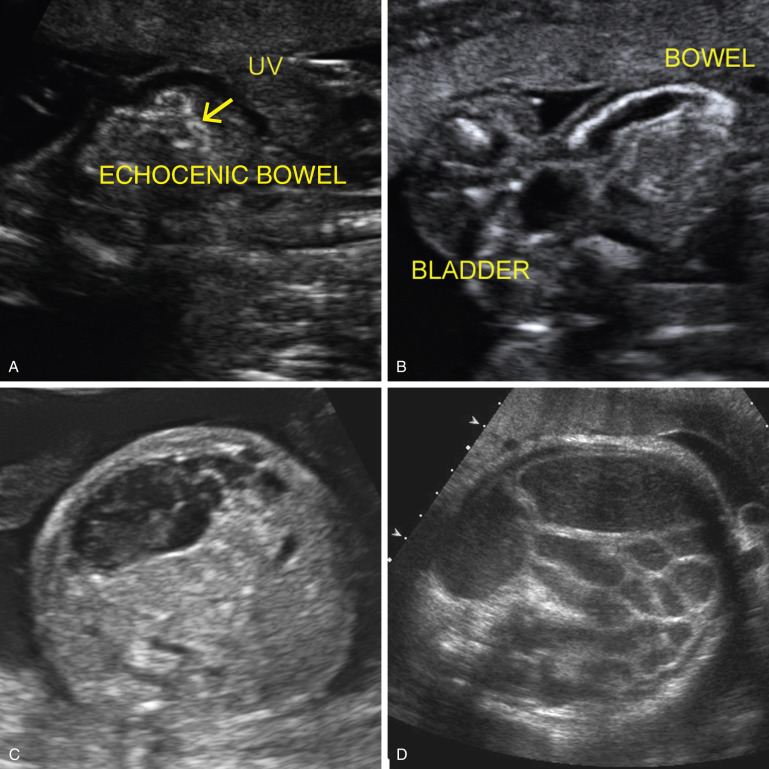
Echogenic small intestine is rarely associated with other anomalies, such as α-thalassemia and intestinal obstruction (see Fig. 14-6 ).
Management of Isolated Echogenic Small Intestine
- 1.
Targeted sonographic evaluation of fetal anatomy, including search for other markers of aneuploidy or evidence of congenital CMV infection, to ensure that the echogenic small intestine is indeed an isolated finding.
- 2.
Evaluation of a priori risk of aneuploidy based on maternal age and previously obtained screening results. If the patient desires, additional prenatal testing for aneuploidy can be offered. Given that the increased risk of aneuploidy is relatively modest and is mainly limited to trisomy 21, cell-free DNA testing is a useful option to consider in this setting.
- 3.
Evaluation of a priori risk of cystic fibrosis based on parental ethnicity and previously obtained carrier screening for CFTR mutations, which should be offered if not previously obtained. Even if the mother screened negative, there is a residual risk that she is a carrier, and that the fetus could be affected. If the prospective parents wish to address this concern, the next step would be to obtain blood from the father of the fetus for carrier screening, and if he tests positive, one or both parents could pursue gene sequencing, which can detect mutations not identified through genotyping panels.
- 4.
Evaluation for congenital infection with maternal serologic tests. Blood is routinely drawn to test for CMV antibodies, both IgG and IgM. To help assess if the infection is acute in the setting of positive IgM antibody, IgG avidity can be determined. Although it is possible to have fetal manifestations from recurrent or secondarily infected CMV, the likelihood is low. Unless there are specific concerns, serologic tests for other infections such as toxoplasmosis may not be warranted. If the maternal serologic tests indicate evidence of acute infection, further evaluation such as amniocentesis for CMV RNA using polymerase chain reaction should be offered.
- 5.
Fetal surveillance in the second half of pregnancy. This typically entails serial ultrasound examinations, often every 4 weeks, to assess growth and amniotic fluid volume. Many institutions also institute weekly or biweekly nonstress tests or biophysical profiles after 32 weeks. Follow-up sonograms will detect most cases with bowel complications.
As a rule, providers taking care of the newborn should always be apprised of antenatal sonographic findings. If the child appears well, no further evaluation is necessary after an in utero finding of isolated echogenic small intestine: follow-up of 48 such infants demonstrated a normal outcome.
Jejunoileal Obstruction
When performing a second trimester obstetric sonogram using a high-frequency transducer, particularly in a thin patient, individual loops of fetal bowel can often be discerned (see Fig. 14-5B ). During the third trimester, fluid can be seen within the bowel lumen; the maximal diameter of normal fetal small intestine is reported to be 7 mm.
The hallmark of distal small bowel obstruction is dilated loops of bowel ( Fig. 14-6D ). This is rarely seen before the third trimester. Polyhydramnios, a dilated stomach, and echogenic bowel are nonspecific findings that are sometimes associated with or are precursors to identification of frank obstruction ( Table 14-5 ).
| Segment | Intestinal Loops | Stomach | Peritoneal Calcification | Ascites | Associated Anomalies | Polyhydramnions |
|---|---|---|---|---|---|---|
| Jejunum | Marked dilation of loops, most of them on the left abdominal side Enlargement of duodenum is common | Large | Frequent | Very rare | Frequent | Frequent |
| Ileum | Usually only minor dilation of a few loops | Normal | Frequent | Frequent | Rare | Rare |
Obstructed fetal small bowel should not be confused with normal fetal colon, which becomes more prominent during gestation, typically appearing as hypoechoic bowel segments, of varying size, at the periphery of the abdomen ( Fig. 14-7A ). The small intestine tends to be more centrally located. Rarely, late in the third trimester, the large intestine can have a characteristic echogenic appearance ( Fig. 14-7B ). This is considered an innocuous finding unless it is detected prior to 36 weeks, when it has been associated with cystinuria.
Etiology
Atresia of a segment of jejunum or ileum is thought to most often be a consequence of vascular disruption leading to ischemic necrosis of a variable length of small bowel. Evidence of swallowed amniotic fluid distal to the atresia suggests that the ischemic event occurred relatively late in gestation. This finding is usually sporadic and the risk of extraintestinal anomalies varies, depending on level of obstruction. In one series of 38 children with jejunal atresia, there were 10 with cystic fibrosis, 4 with congenital heart disease, and 5 with a variety of other defects including one with Down syndrome. In contrast, only 1 of 45 children with ileal atresia had an associated anomaly. Other abnormalities of the gastrointestinal tract, such as esophageal atresia, duodenal obstruction, and anorectal malformation, are sometimes seen in association with jejunoileal obstruction.
Some rare autosomal recessive syndromes are associated with jejunoileal atresia: “apple peel” atresia is associated with an extensive atretic segment and therefore has a guarded prognosis, and another syndrome with multiple atretic segments has a poor prognosis. Midgut volvulus, with or without the whirlpool sign, has also been reported prenatally. Meconium ileus, which is sometimes due to cystic fibrosis, is a common cause of fetal small bowel obstruction. This occurs from an accumulation of inspissated meconium in the distal ileum.
Evaluation and Antenatal Management
The differential diagnosis includes congenital intestinal pseudo-obstruction, colonic atresia, Hirschsprung disease, and congenital chloride diarrhea. Hydroureter should be reasonably easy to distinguish from dilated bowel by ultrasound imaging as it can be traced to the bladder or kidney. In some studies, fetal MRI has been reported to be useful.
Fetal small bowel obstruction has been associated with a high incidence of prematurity and growth restriction, possibly related to an inability to obtain oral nourishment from the amniotic fluid.
Neonatal Management
Treatment for jejunoileal atresia is surgical, and the prognosis is excellent unless there are multiple sites of atresia or extensive “apple peel” atresia. The treatment for volvulus is also surgical and prognosis depends on the length and condition of the affected bowel; “short gut” syndrome is the major cause of long-term morbidity.
Initial treatment for simple meconium ileus is one or more isotonic water-soluble contrast enemas, which is successful in the majority of cases.
Meconium Peritonitis
Meconium peritonitis is thought to result from inflammation that occurs in response to intestinal rupture. The perforation can be due to intestinal atresia, volvulus, intussusception, internal hernia, or vascular compromise; often the cause is unknown. In the past, pediatric series of symptomatic newborns reported high rates of morbidity and mortality but the prognosis of sonographically detected prenatal cases is much better; in two recent papers, there were 5 deaths out of a total of 107 cases.
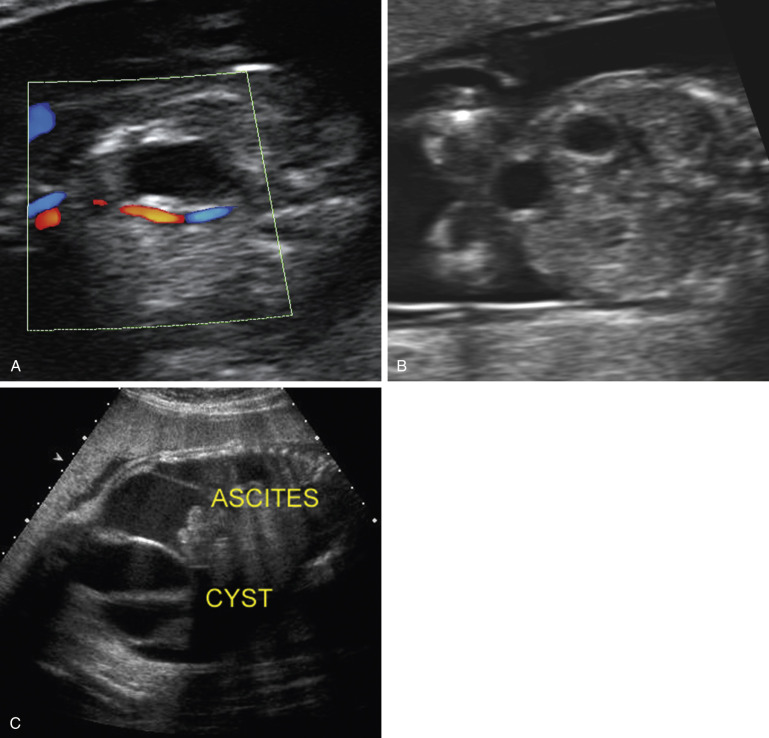
Meconium peritonitis is suspected when extraluminal abdominal calcifications and ascites are seen. Extraluminal calcifications form along the visceral or parietal peritoneum, often on the surface of the liver or under the diaphragm, and appear as linear echogenic densities possibly with posterior acoustic shadowing ( Fig. 14-8A ). These changes may also extend through the patent processus vaginalis into the scrotum ( Fig. 14-8B ). Ascites may be observed initially and then noted to resolve over time.
A meconium pseudocyst occurs when extravasated intestinal contents become walled off. It typically appears as a complex, round intra-abdominal cystic collection with an echogenic wall ( Fig. 14-6C ).
Several methods of grading the severity of meconium peritonitis have been suggested; all agree that the greater the severity, the greater the likelihood that the newborn will be symptomatic. Using one such classification system, when sonographic abnormalities included only intraperitoneal calcifications, 0/18 infants required surgical intervention; but when there were additional findings such as ascites, pseudocyst, bowel dilation, or polyhydramnios the likelihood of surgical intervention increased. The authors suggest that it may be safe to deliver a fetus with only scattered intraperitoneal calcifications in a community hospital, but when other sonographic findings are noted, delivery in a tertiary care center is probably warranted.
Pediatric series have demonstrated a strong association between meconium peritonitis and cystic fibrosis. Interestingly, cases associated with cystic fibrosis are less likely to demonstrate intraperitoneal calcifications, perhaps because the intestinal contents are less inflammatory or remain more localized. Genetic testing for cystic fibrosis is routinely offered early in pregnancy, and results of such testing should be evaluated if meconium peritonitis is identified. Other factors that have been reported in association with meconium peritonitis include parvovirus infection and hepatitis A.
Rectum
Anorectal Malformations
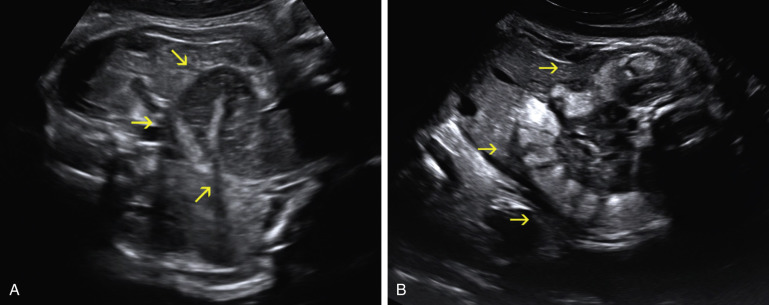
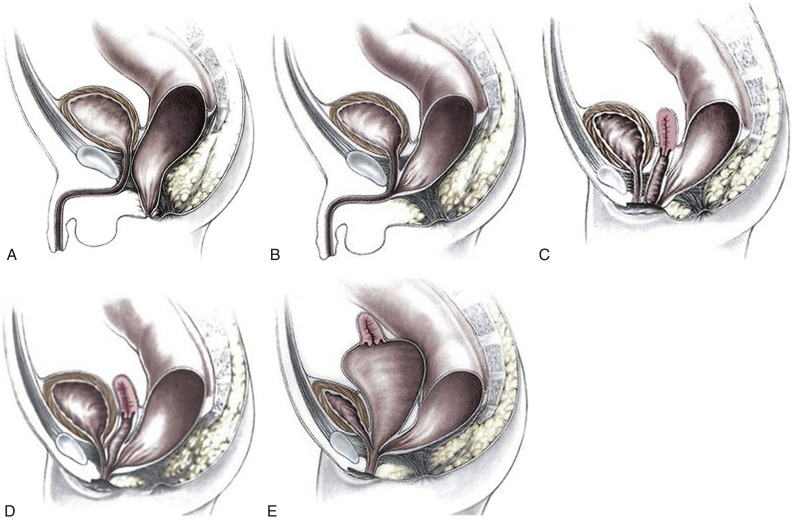
Anorectal malformations result from abnormal partitioning of the cloaca by the urorectal septum into the urogenital sinus anteriorly and the rectum posteriorly ( Fig. 14-9 ); these malformations are also referred to as urogenital sinus malformations . Earlier failure of development of the urorectal septum results in a higher, more severe abnormality. The mildest defects are often referred to as anal atresia or imperforate anus and often involve a rectal fistula that opens anterior to the anal muscular complex ( Fig. 14-10A through C ). Anorectal malformation without fistula, which is strongly associated with Down syndrome, accounts for only 4% of anorectal malformations. In females, the most severe abnormality is persistent cloaca ( Fig. 14-10D ), which will be discussed separately.
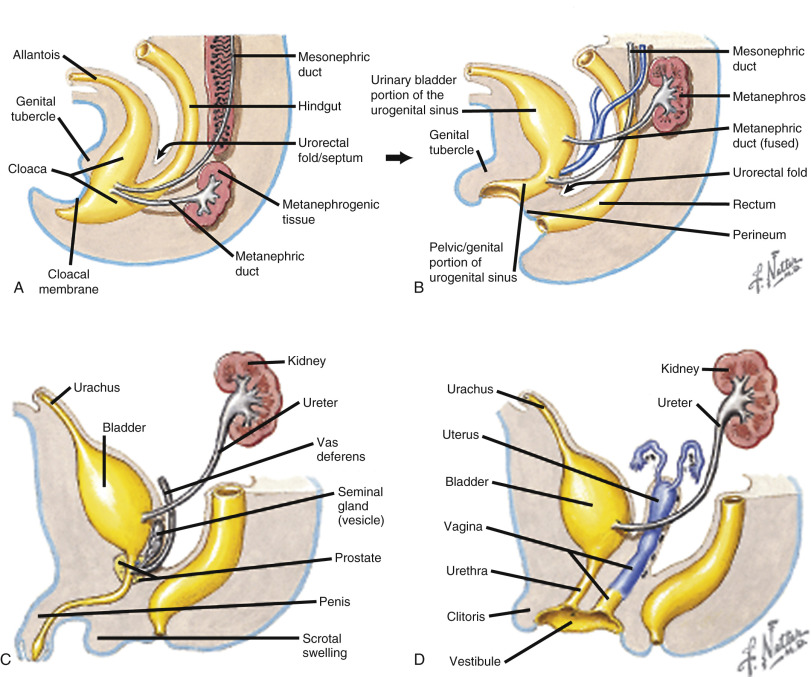
Prevalence and Associated Anomalies
The EUROCAT survey of 4.6 million births identified 1846 cases of anal anomalies, with a prevalence of 4 per 10,000 births. Fortunately lower lesions are far more common than higher lesions; only 10% of anal atresias were above the level of the levator ani. A wide variety of additional findings are present in the majority of cases and include VACTERL (see Table 14-3 ), chromosomal abnormalities, other known syndromes and sequences, and multiple anomalies involving the genital system, central nervous system, face, limbs, musculoskeletal system, and to a lesser extent, all other organ systems.
Although most cases are sporadic, like some other abnormalities, there is often a genetic component, and anorectal malformations are associated with a variety of single gene syndromes. There are also reports of familial nonsyndromic anorectal malformations.
Sonographic Diagnosis
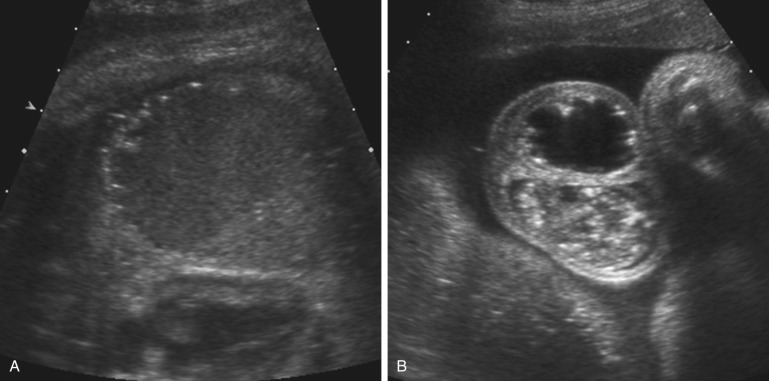
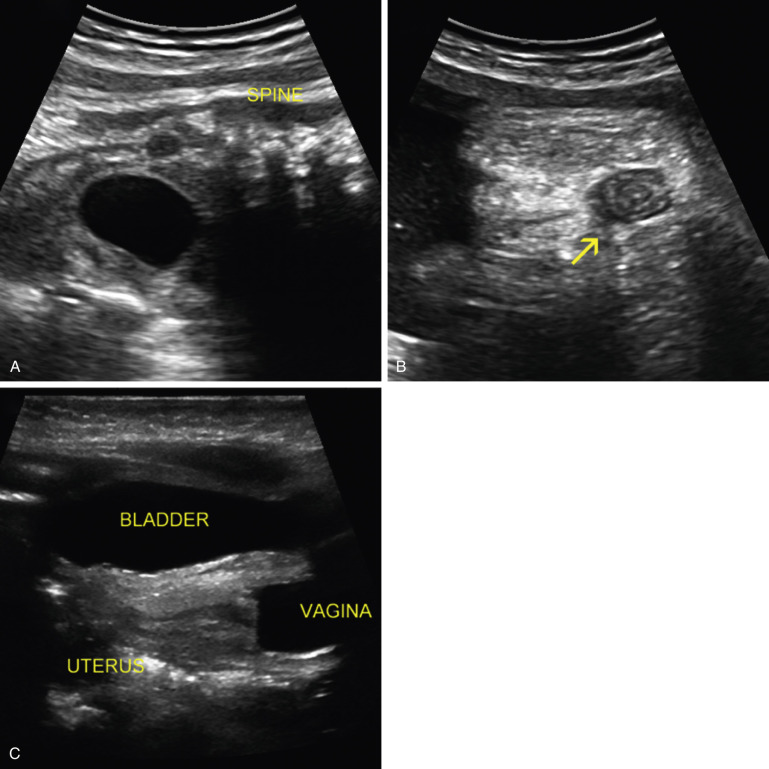
Cystic fluid collections in the first trimester have been associated with anal atresia ( Fig. 14-11A and B ). In the second trimester, observation of a dilated rectum allowed for the prenatal sonographic diagnosis in only 11 of 69 (16%) cases of anal atresia and in only 3 cases when it was isolated. Calcified intraluminal meconium is sometimes seen in cases of anorectal malformations, but it is not known how often this occurs. These findings are likely related to the presence of an enterourinary fistula, which allows urine to distend the rectum and interact with meconium, resulting in a calcified appearance. In contrast, one might not see any sonographic findings in the case of rectoperineal or rectovestibular fistulas (see Fig. 14-10A and C ) or of anal atresia without a fistula.
More recent studies address the possibility of direct visualization of the fetal anal sphincter as a circular hypoechoic area with a small echogenic center ( Fig. 14-12B ), which can be identified in more than 90% of fetuses after 23 weeks’ gestation. In one study, inability to image this structure allowed for the prenatal diagnosis of 17 cases of anal atresia. Some groups utilize three-dimensional (3D) sonography for this purpose. A combination of 2D and 3D techniques was used to evaluate 189 fetuses at increased risk for anorectal malformation based on the presence of one or more other congenital structural anomalies known to be associated with anorectal malformations. The investigators correctly excluded anorectal malformation in 175 fetuses and correctly detected an abnormality of the anus in the other 14.
Persistent Cloaca
This most severe form of anorectal malformation is found only in females. It is very uncommon, with a prevalence of only 0.2 per 10,000. With persistent cloaca, the distal portion of the urinary tract, vagina, and rectum all lead to a single common channel that opens onto the perineum (see Fig. 14-10D ). In 30% of cases, hydrocolpos develops as a result of passage of urine and meconium into the vagina, which is more compliant than the bladder or rectum ( Fig. 14-10E ). Development of hydrocolpos can result in several additional pathologic changes, some of which can be seen by ultrasound imaging in the third trimester ( Figs. 14-11 and 14-13 ).
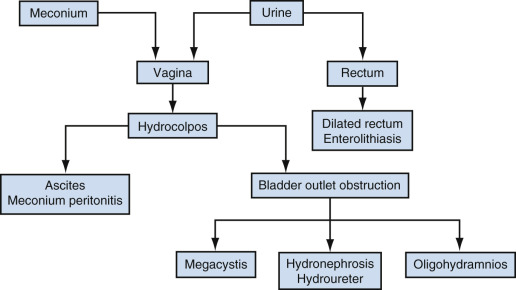
Hydrocolpos presents as a midline cystic mass in the pelvis, posterior to the bladder, extending to the perineum ( Fig. 14-11C ). There is often a longitudinal vaginal septum, and there may be fluid/fluid levels due to mixture of urine and meconium. The contents of the cystic mass will not be uniformly sonolucent as is the case in megacystis.
Hydrocolpos is frequently associated with urinary tract obstruction; hydronephrosis and oligohydramnios were the next most common abnormalities seen on prenatal ultrasound imaging. If obstruction is at the level of the bladder outlet, there will be megacystis; if obstruction is at the level of the trigone, the bladder will appear normal or small.
Ascites is seen in a minority of cases. It is thought that this occurs as a result of retrograde passage of cloacal contents through the fallopian tubes. Because of the inflammatory response triggered by this material, abdominal calcifications characteristic of meconium peritonitis may be seen. In some cases, dilated loops of bowel will demonstrate intraluminal calcifications due to mixing of meconium with urine. Additional findings that may be seen in this setting include multicystic dysplastic kidney, ectopic/malpositioned kidney, single umbilical artery, and spinal abnormalities.
The prenatal diagnosis of persistent cloaca is challenging, and in two recent series was made in only 6% of cases. This is likely related to the rarity of this condition, the complex and variable nature of the ultrasound findings with hydrocolpos present in less than half. When this diagnosis is suspected, fetal MRI is often useful. On a sagittal view of a normal third trimester fetus, the rectum should extend at least 10 mm below the bladder; in 5 of 6 fetuses with persistent cloaca, third trimester MRI demonstrated a high and dilated rectum.
Neonatal Management
Examination of the perineum of the neonate will often establish the diagnosis. In males, waiting 24 hours is suggested so that intraluminal pressure in the rectum can increase sufficiently to allow passage of meconium through a fistula, either on the perineum or in the urine. In females, inspection of the perineum can demonstrate a single orifice, signifying the presence of a cloaca, or reveal a fistula in the vestibule or on the perineum. Careful evaluation for associated anomalies by physical examination and imaging is also important.
Definitive surgical repair depends upon the level of the lesion. A low lesion with a fistula on the perineum can undergo primary repair, whereas a higher fistula often requires placement of a colostomy followed by a more complex surgical procedure.
Liver, Spleen, and Gallbladder
Liver
The umbilical vein enters the lower abdomen and travels in a cephalad and posterior direction to enter the liver. Within the liver, the largest branch curves toward the right, away from the stomach, where it connects with the ductus venosus and drains into the inferior vena cava just below the right atrium. In the transverse plane, at the level where abdominal circumference is measured, the vein is a prominent C-shaped structure within the liver, curving away from the stomach bubble (see Fig. 14-2A ).
Occasionally the umbilical vein is noted to curve toward the left side of the fetal abdomen, toward the stomach ( Fig. 14-14A ). This represents persistence of the right umbilical vein , which may be associated with other anomalies but when isolated is considered a normal variant.
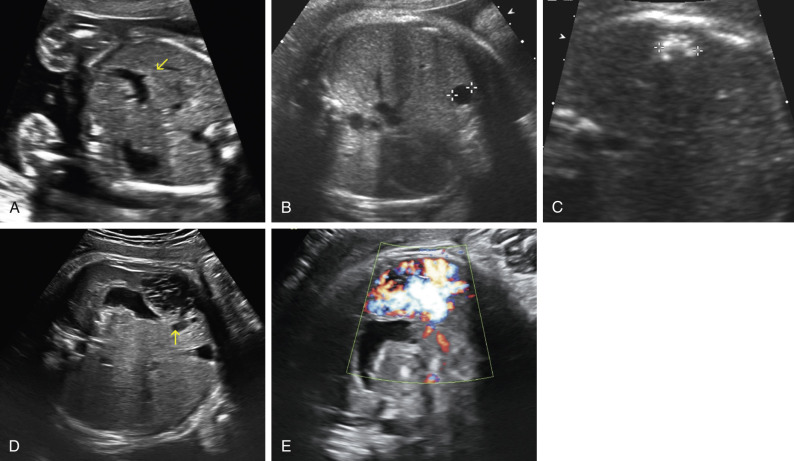
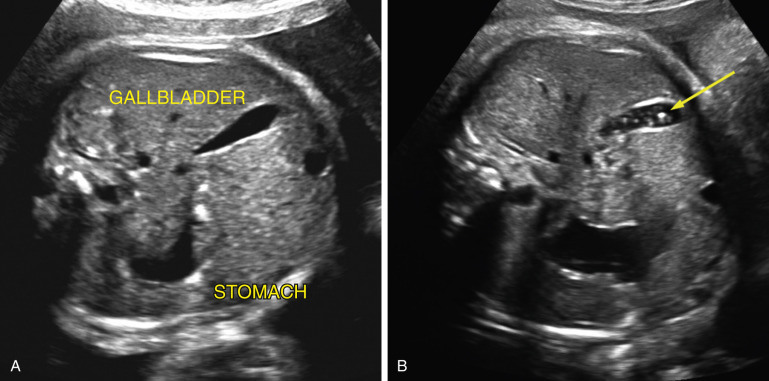
Choledochal cysts result from dilation of the biliary tract, most often fusiform dilation of the common bile duct ( Fig. 14-15 ). They present as sonolucent cysts at the hepatic hilum. The differential diagnosis includes benign hepatic cyst ( Fig. 14-14B ). The diagnosis is confirmed after birth by ultrasound imaging and magnetic resonance cholangiopancreatography. Treatment requires surgical excision and biliary-enteric anastomosis (hepatojejunostomy or hepatoduodenostomy) to prevent obstructive jaundice, cholangitis, pancreatitis, liver fibrosis, and malignancy; the timing of surgery is controversial.

Hepatic calcifications/echogenic foci in the liver parenchyma ( Fig. 14-14C ) should be distinguished from calcifications along the surface of the liver from meconium peritonitis (see Fig. 14-8A ) and from echogenic material within the gallbladder ( Fig. 14-16B ). Hepatic parenchymal calcifications are relatively common; the prevalence may be as high as 1 : 1000. In the largest reported series, 40 of 61 cases had additional sonographic abnormalities; of these, there were 10 cases with aneuploidy and 1 with congenital CMV infection. Outcomes were better for the 21 cases with isolated hepatic calcifications: 1 fetus had parvovirus and 1 had trisomy 21, but the others fared well. In a review of the four largest series, 93% of 63 cases with isolated intrahepatic echogenic foci had normal outcomes compared with 39% of 54 cases when there were other ultrasound findings.
Related posts:
 Ultrasound Evaluation of the Fetal Central Nervous System
Ultrasound Evaluation of the Fetal Central Nervous System
 Measurements Frequently Used to Estimate Gestational Age and Fetal Biometry
Measurements Frequently Used to Estimate Gestational Age and Fetal Biometry
 Role of Doppler Sonography in Obstetrics
Medications and Reported Associated Malformations
Role of Doppler Sonography in Obstetrics
Medications and Reported Associated Malformations
 Role of Sonography in Gynecologic Interventions
Role of Sonography in Gynecologic Interventions
 Ultrasound Features of Fetal Syndromes
Ultrasound Features of Fetal Syndromes
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


