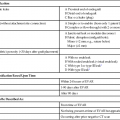
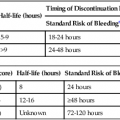
Vasoactive agents are commonly employed in a variety of settings in the hospitalized patient, including the interventional radiology suite. Safe and effective pharmacologic manipulation of an individual patient’s hemodynamics for therapeutic or diagnostic purposes requires a thorough understanding of patient-specific factors and a firm comprehension of the pharmacology of the agents employed. In this chapter we review the pharmacology, hemodynamic impact, and intraarterial use (where indicated) of selected vasoactive agents that may be used in a variety of clinical settings in the interventional radiology suite. The reader is referred to Chapter 16 for more detail regarding the specific use of these agents in the setting of medical emergencies. α1-Adrenergic receptors are G protein–coupled transmembrane receptors whose intracellular signaling is through membrane phospholipids; they comprise three subtypes: 1a, 1b, and 1d. The subtypes have wide but variable distribution, with the highest expression for la being found in the liver and lower levels in the heart, cerebellum, and cerebral cortex. The lb subtype shows up greatest in the spleen, kidney, and cerebellum, whereas the 1d subtype has its greatest expression in the cerebral cortex and aorta.1 Subtype-specific agonists and antagonists exist for these receptors but are for research purposes only. Clinically available agonists and antagonists are nonspecific in terms of subtype binding. Activation of these receptors by the clinically relevant agents leads to smooth muscle contraction that, in the vasculature, results in vasoconstriction. α2-Adrenergic receptors are transmembrane receptors whose intracellular signaling is mediated through G-protein inhibition of adenylate cyclase. The three subtypes—2a, 2b, and 2c—are widely distributed and each contribute uniquely though not completely for the various responses seen with α2 agonists. The α2b subtype has been demonstrated to be responsible for the short-term hypertensive response that may occur with α2 agonists, whereas the α2a subtype has been implicated in the sympatholytic response and elements of the anesthetic response to these agents. Currently there are no subtype selective α2 agonists, and all α2 agonists (clonidine and dexmedetomidine) are nonspecific in their action. Lower doses of these agents tend to produce hypotension through sympatholysis, whereas at higher doses the α2b receptor present on the smooth muscle cells of resistance vessels predominates, leading to a hypertensive response.2 The β receptors are subdivided into β1, β2, and β3 subtypes, and all generally use G protein–mediated activation of adenylyl cyclase as their intracellular signaling pathway. They have wide distribution and divergent clinical responses despite their biochemical relationship. β1-Adrenoreceptors are predominantly found in the heart, whereas β2 receptors are present in the smooth muscle beds of the lung, peripheral vasculature, and uterus. β3-Adrenergic receptors are unique in that one of their roles appears to be in metabolic regulation.3 Activation of β1 receptors is primarily responsible for increased inotropy and chronotropy in the heart. Other actions mediated by these receptors include release of renin from the kidneys and activation of lipolysis.4 Their effect on cardiac contractility and heart rate results in increased cardiac output and sometimes blood pressure (BP), although this latter effect is often clinically blunted or even reversed by agonism at the β2 receptor. Although β1-specific antagonists (e.g., esmolol) are clinically available, there are no β1-specific agonists (although there are some fairly β2-specific agents, primarily used to treat bronchospasm). These improvements in cardiac output come at a price, however, because the increased contractility and heart rate significantly increase myocardial oxygen demand while reducing supply, putting the heart at ischemic risk. Long-term overstimulation of these receptors through humoral feedback mechanisms has been implicated in the molecular pathogenesis of chronic congestive heart failure (CHF), and strategies to interrupt these processes using β1 antagonists (β-blockers) have led to some clinical success.5 β3-Adrenergic receptors were originally thought to have little to no clinical relevance in terms of hemodynamics. Their primary role was believed to be in the metabolic regulation of energy metabolism via effects on adipose tissue. Although their ultimate place in terms of physiologic effects is still a matter of investigation, other mechanisms attributed to this subtype include gastric acid secretion, coupling to potassium channels in smooth muscle, and interactions with nitric oxide–dependent vasorelaxation.6–8 Despite these possibilities, from a clinical point of view, the β3 receptor does not yet have a defined role in the immediate clinical response to the commonly used vasopressors and inotropes. A separate group of receptors involved in vasomotor tone are the dopamine receptors. Although they are not usually classified as adrenergic receptors, their primary agonist is the catecholamine dopamine. Like the adrenergic receptors, these are G protein–coupled transmembrane receptors. Dopamine receptors have two different nomenclature systems depending on whether they are in the central nervous system (CNS) or the peripheral vasculature. Those in the CNS were originally classified into two types, the D1 and D2 subtypes, based on their activation or inhibition of adenylate cyclase. Modern cloning techniques have further defined them into two superfamilies: the D1-like group that includes the D1 and D5 subtypes and the D2-like group that includes the D2, D3, and D4 subtypes.9 The DA2 receptor subtype is similar to the CNS D2-like receptors and is mostly found on the presynaptic adrenergic nerve terminals and in the sympathetic ganglia.9 Agonism at these receptors leads indirectly to vasodilation by inhibiting the release of norepinephrine at the adrenergic nerve termini and by suppressing transmission at the sympathetic ganglia, leading to an overall drop in sympathetic activity. Agents used to sustain BP in hypotensive patients can work through several mechanisms but usually exert their action via stimulation of adrenergic receptors. The medications discussed in this section can have very different pharmacologic actions based on their receptor-activity relationship. The BP elevation is due to either an increase in systemic vascular resistance or an increased cardiac contractility, resulting in increased cardiac output, or a combination of both. These differences are discussed individually within each drug section. These pharmacologic agents generally have the same adverse event profile, although there are nuances depending on the receptor-activity relationship. Agents that are potent α1 agonists can produce excessive vasoconstriction and necrosis to poorly perfused tissues (especially in hypovolemic or under-resuscitated patients). Myocardial ischemia and dysrhythmias can occur due to β1 stimulation. Other potential adverse effects that can occur are metabolic derangements (e.g., lactic acidosis) and soft-tissue damage from extravasation.10–13 When the decision is being made to administer vasopressors, fluid status should always be assessed to truly determine the cause of the patient’s hypotension. Phenylephrine is a short-acting adrenergic agonist that is highly specific for the postsynaptic α1 adrenoreceptor and has slight activity at the presynaptic α2 adrenoreceptor.14 This activity results in an overall increase in vascular tone, as demonstrated by an increase in systemic vascular resistance when a pulmonary artery catheter is in place. Owing to its lack of cardiac stimulation, phenylephrine has been used as a primary vasopressor in patients in septic shock who require hemodynamic support. It has been shown in multiple studies to have little effect on important cardiac indices, including cardiac index, in noncardiac patients. Adverse effects on cardiac performance have been shown in patients with cardiac dysfunction, resulting in decreased cardiac performance due to an increase in peripheral resistance.15,16 Phenylephrine can be dosed either as an intravenous (IV) bolus or a continuous infusion. For IV bolus administration, phenylephrine can be diluted to 100 µg/mL in a syringe, with 1 to 2 mL (100-200 µg) administered at a time, and doses given to achieve a goal mean arterial pressure (MAP) greater than 65 mmHg. This administration technique has been used in both obstetric patients and patients undergoing anesthetic induction during short procedures.17,18 Phenylephrine IV infusions are more logical for patients who require hemodynamic support for prolonged periods. Doses for infusions are weight based and typically range from 0.1 to 4 µg/kg/min. Phenylephrine has been associated with decreased cardiac performance and increased splanchnic vasoconstriction (compared with norepinephrine), which limits its clinical usefulness.19 For this reason, it is no longer considered first-line therapy for vasopressor-dependent septic shock.20 Phenylephrine can be considered useful in the management of hypotension associated with anesthesia induction (including spinal), and for the short-term management of hypotension not due to sepsis. Ephedrine is a mixed adrenoreceptor agonist. It has effects on both the α1 and β1 receptors in the cardiovascular system, resulting in vasoconstriction and tachycardia. It also has an indirect effect, causing postganglionic release of norepinephrine.21–24 Ephedrine is effective with bolus dosing in preventing and treating epidural or spinal anesthesia–induced hypotension, with doses ranging from 10 to 50 mg intravenously or intramuscularly.25–29 IV administration results in immediate effects that are short acting, whereas doses given intramuscularly may not take effect for 10 to 20 minutes, with a duration of 60 minutes. Continuous infusions of ephedrine have been thought to deplete norepinephrine stores, resulting in tachyphylaxis, but this is debatable.23,24 Ephedrine is also known to possess stimulant, hallucinogenic, and euphoric properties that have resulted in its use being limited to short-term procedural use in obstetric and surgical/procedural settings.30,31 Norepinephrine is a potent α1 and α2 agonist and, to a lesser extent, a β1 and β2 agonist. Its primary effect is a profound increase in systemic vascular resistance through vasoconstriction of vascular beds. Whereas this is similar to phenylephrine, the β1/β2 stimulation supports cardiac function in the presence of increased resistance, so its effects on cardiac function range from no change to slight increase in cardiac output.10,32 Recent published guidelines have promoted the use of norepinephrine or dopamine as the initial vasopressor in management of septic shock, which is a shift from previous clinical practice in which norepinephrine was only used in dopamine-resistant septic shock.20,33 Norepinephrine may be preferable to dopamine in patients in whom β1 stimulation would be deleterious, because norepinephrine possesses more potent α1 and less potent β1 activity. Excessive vasoconstriction due to the potent α1 stimulation can occur, resulting in regional ischemia (skin, splanchnic perfusion). Norepinephrine is dosed as a continuous IV infusion, with doses in clinical trials ranging from 0.1 to 2.2 µg/kg/min.34 The hemodynamic effects one can expect are increases in systemic vascular resistance (and ultimately MAP), no to mild increase in cardiac output/cardiac index, and slight increases in heart rate. Norepinephrine should be used as a first-line vasopressor in patients with septic shock, who are at higher risk for adverse outcomes associated with arrhythmias. Norepinephrine should also be used in the presence of phenylephrine-resistant hypotension because it is a more potent α1 agonist. Dopamine is a vasoactive agent that has multiple receptor binding sites throughout the body, with varying dose-response activity at dopamine, β1, β2, and α1 receptors (Table 23-1), although it has been shown that there is a degree of overlap in critically ill patients beginning at doses as low as 3 µg/kg/min.35 The high doses of dopamine needed to maintain BP (15-20 µg/kg/min) can lead to tachyarrhythmias, limiting its usefulness as a first-line agent in managing shock. It can also cause disturbances in pulmonary function, including increases in pulmonary capillary wedge pressure, shunting, and decreased oxygen pressures.36 Dopamine has also been shown to increase splanchnic and mesenteric ischemia.11,37 Overall, norepinephrine has become the vasopressor of choice in the management of septic shock.20,38 Low-dose dopamine (<3 µg/kg/min) had been thought to promote renal blood flow and increase renal function, especially in the critically ill, who are at increased risk for acute renal failure. Multiple studies have now shown that this management strategy has no effect on renal or survival outcomes and should be abandoned.39,40 TABLE 23-1 Comparison of Relative Receptor Activity of Adrenergic Agonists
Vasoactive Agents
Adrenergic Receptor Pharmacology
Agents Used to Increase Blood Pressure
Phenylephrine
Ephedrine
Norepinephrine
Dopamine
Agent
α1
α2
β1
β2
DA
Dobutamine
+
0
++++
++
0
Dopamine:
0-3 µg/kg/min
0
0
+
0
++++
3-10 µg/kg/min
0/+
0
++++
++
++++
10-20 µg/kg/min
+++
0
++++
+
0
Epinephrine:
<0.5 µg/kg/min
++
++
++++
+++
0
>0.5 µg/kg/min
++++
++++
+++
+
0
Norepinephrine
Related posts:
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree

 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access