Abstract
Dilatation of the lateral ventricles of 10 mm or more is the most frequent sign of possible abnormality detected on antenatal ultrasound. It is not a disease but rather a sign that represents a common endpoint of various pathologic processes with different outcomes and prognosis. It can be a benign finding, but it can also be associated with such conditions as aneuploidies, congenital infections, cerebral vascular accidents, and other fetal cerebral or extracerebral abnormalities. Depending on the cause, there are different implications for long-term neurodevelopmental outcome. Apart from the underlying cause, the prognosis depends on coexistence of other anomalies and eventual progression of ventricular dilatation. Investigations therefore focus on determining a cause, excluding coexisting anomalies, and following up to assess for progression. Counseling parents whose fetus has ventriculomegaly should reflect these challenges and uncertainties.
Keywords
ultrasound, ventriculomegaly, MRI, management, outcome, neurodevelopment, chromosomal abnormality, congenital infection
Introduction
Ventriculomegaly (VM) is a nonspecific term, describing dilatation of the lateral cerebral ventricles, usually defined as greater than 10 mm at the level of the atria. VM is the most frequent abnormal central nervous system (CNS) finding detected with prenatal imaging techniques. It is not a disease, but rather a sign that represents a common endpoint of various pathologic processes with different outcomes and prognosis; it may also be a physiologic finding representing an extreme normal variation.
The prognosis depends to a large degree on the underlying cause, the coexistence of other anomalies, and eventual progression. Because these cannot always be determined with confidence, counseling for VM can be difficult. It can be a benign finding, but it can also be associated with aneuploidies, congenital infections, cerebral vascular accidents or hemorrhage, and other fetal cerebral or extracerebral abnormalities, with different implications for long-term neurodevelopmental outcome.
Disease
Definition
Fetal VM is an ultrasound (US) finding that refers to dilatation of the lateral ventricles of the brain, typically defined as greater than 10 mm at the level of the atria, with or without dilatation of the third or fourth ventricles. In the pediatric literature, the term hydrocephalus usually refers to expansion of the ventricular system associated with increased cerebrospinal fluid (CSF) pressure, secondary to a disturbance in the dynamics of CSF circulation. Because it is impossible to measure CSF pressure in utero prenatally, this condition is preferably referred to as VM.
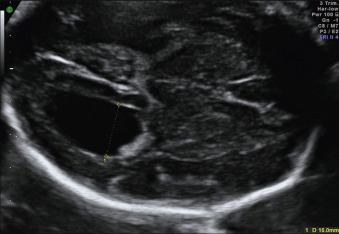
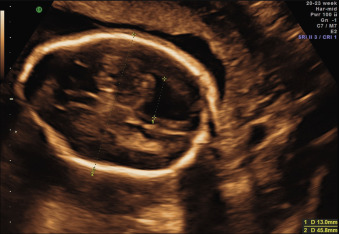
The ventricular atrial width is roughly constant after 15 weeks’ gestation. VM is defined as a width at the level of the atria of the lateral ventricle greater than 10 mm; this corresponds to about 4 standard deviations above the mean of 7.6 mm. Most authors consider an atrial width of less than 10 mm normal, a width between 10.0 and 15.0 mild VM, and a width greater than 15 mm severe VM. However, definitions vary in the literature. The terms mild, moderate, and severe VM for atrial widths 10 to 12 mm, 13 to 15 mm, and greater than 15 mm are also often used ( Figs. 43.1 and 43.2 ).
Prevalence and Epidemiology
Because the incidence of VM depends on the definition used, it has been reported to range from 1 : 50 to 1 : 1600. The incidence of severe VM is about 1 : 1000 newborns. Discrepancies in reported incidences are also due to differences in measuring techniques or gestational age at examination, assessment of only one hemisphere, a lack of precise correlation in the diagnosis between prenatal and neonatal life, and the possible prenatal “resolution” of the condition. Male fetuses are diagnosed with VM slightly more often than female fetuses.
Etiology and Pathophysiology
Many cases of VM are due to an imbalance between production and absorption of CSF caused by an obstructive event. CSF is formed mainly at the level of the choroid plexus and flows from the lateral ventricles through the foramina of Monro into the third ventricle, and then via the aqueduct of Sylvius into the fourth ventricle. At this level, it passes through the foramen of Magendie (in the roof of the fourth ventricle), or through the foramina of Luschka (at the sides of the fourth ventricle) into the subarachnoid space around the cerebral hemispheres. From there, CSF is reabsorbed by the arachnoid granulations that are mainly distributed along the superior sagittal sinus. If there is obstruction in the ventricular system, VM is defined as noncommunicating or obstructive . The obstruction can occur at different levels of the ventricular system, and different etiologies are known—either congenital, or secondary to intrinsic or extrinsic lesions. Generally, the more distal the obstruction is from the foramina of Monro, the greater the degree of the involvement of the ventricular system (see Fig. 43.2 ).
Noncommunicating VM includes the unilateral and bilateral obstruction of foramina of Monro, congenital narrowing of the aqueduct of Sylvius, aqueductal forking, or the presence of a septum within the aqueduct. Some cases of aqueductal stenosis may be inherited as an X-linked recessive trait or, more rarely, as an autosomal recessive disease.
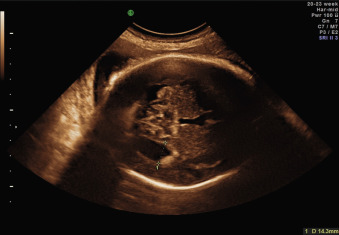
Ventricular obstruction can also be caused by intrinsic processes such as inflammation, secondary to infections such as cytomegalovirus and toxoplasmosis ( Fig. 43.3 ); by hemorrhage; or by other extrinsic events such as tumors, arachnoid cysts, or malformations of the CNS that impair the normal circulation of CSF between the ventricular and the extraventricular system. Open neural tube defects and posterior fossa abnormalities are also often associated with VM ( Fig. 43.4 ).
The term communicating VM describes conditions such as sagittal sinus thrombosis, in which the cause is outside the ventricular system and a communication with at least part of the subarachnoid space is possible, or conditions with increased CSF production, such as choroid plexus papilloma without an obvious obstruction. VM can also develop without obstruction, in prosencephalic developmental anomalies (e.g., holoprosencephaly, agenesis of the corpus callosum [see Fig. 43.5 ]) and disorders of neural proliferation (e.g., microcephaly, megalencephaly) or neuronal migration (e.g., lissencephaly, schizencephaly) ( Chapter 34 , Chapter 36 , Chapter 39 ). Finally, VM can be the consequence of acquired destructive processes of the brain, resulting from vascular accidents or infections ( Chapter 40 ).
Manifestations of Disease
Clinical Presentation
Clinical presentation depends on the causes and the presence of intracranial and extracranial associated anomalies. VM is commonly defined as isolated only if there is no US evidence of intracranial or extracranial associated malformations or other markers of aneuploidy at the time of initial presentation (see Fig. 43.1 ).
Prenatal presentation is usually at the time of the routine second-trimester US scan. Detailed expert neurosonography is indicated to search for possible causes, and further targeted investigations such as fetal brain magnetic resonance imaging (MRI), screening for congenital infection, or screening for antiplatelet antibodies may be indicated.
The most important prognostic factors in VM are
- •
association with other anomalies,
- •
the true cause of the condition, and
- •
the progression of ventricular dilatation.
Mild isolated VM ( Fig. 43.1 ) is usually asymptomatic at birth. Severe VM can manifest with signs of increased CSF pressure, including sutural diastasis, dilatations of scalp veins, bulging of the anterior fontanelle, downward deviations of the eyes, irritability, abducens nerve palsy, and, in advanced cases, bradycardia, and bradypnea. Perinatal mortality is higher in cases of severe VM compared with mild VM.
Assessing the neurodevelopmental performance of children with a prenatal diagnosis of isolated mild ventriculomegaly is challenging. Wide variation exists in the reported prevalence of neurodevelopmental delay in infants with this condition. Part of the difficulty in providing an accurate prognosis and outcome in these infants may arise from methodologic problems, as mentioned earlier. The retrospective nature of most studies, small heterogeneous samples, inconsistent imaging, and different types of follow-up have likely contributed to these variable results. Furthermore, the term neurodevelopmental outcome per se can be inappropriate when dealing with subtle brain anomalies such as VM, because it includes a wide spectrum of signs that are not always easily measured, and that represent a continuous interaction between pathologic, environmental, and adaptive factors. The optimal time to assess neurodevelopmental outcome in children with isolated mild VM is also debatable. Very early assessment may not accurately predict neurodevelopmental outcomes during later life; in contrast late evaluations may be biased by the influence of socioeconomic, parenting, environmental, and educational factors, which may significantly affect developmental measures, especially when looking for subtle differences. Control populations represent another peculiar issue: defining a neurodevelopmental measure as abnormal implies knowledge of how often the measure is abnormal in the nonaffected population. However, the prevalence of neuropsychologic morbidity in the general population, defined as not being affected by brain anomalies, is extremely variable and it has been reported to be as high as 7% to 10% in some studies.
A peculiar example of how interpretation of such a subtle brain finding can change in relation to the time and type of assessment is provided by the study by Atad-Rapoport, who looked at the neurodevelopmental performance of children with isolated unilateral VM and asymmetric ventricles at school age. In a previous study, the authors had found that children with unilateral mild ventriculomegaly scored significantly lower on mental development and behavioral rating scales, and that about 20% of children with asymmetric ventriculomegaly showed developmental delay when evaluated at a preschool age. These findings completely changed when these children were evaluated at school age, when their neuropsychologic profile did not significantly differ from that of the general population for most of the observed outcome measures, although the study included only a part of the original cohort.
Finally, the meaning of neuropsychologic impact after fetal ventriculomegaly is another fundamental issue; the large majority of the studies dealing with neurodevelopmental outcome in isolated mild VM do not discriminate between “neurodevelopmental delay” and “neurologic disability,” and do not quantify the impact of an abnormal neuropsychologic outcome on daily quality of life. Mild delay may not be always associated with disabilities and may have only a minor impact on the overall quality of life; on the other hand, severe delay is commonly associated with disabilities that can significantly affect quality of life.
Bearing in mind all these limitations, the prevalence of abnormal neurodevelopmental outcome in fetuses diagnosed with isolated mild VM confirmed at birth has been reported to be about 8% in a recent systematic review. Some studies have focused on the finding of unilateral or asymmetric VM, defined as a difference of width of 2 mm or more in ventricular size between the two hemispheres, to assess outcomes.
The incidence of neurodevelopmental delay in cases of unilateral mild VM has been reported to be about 7%—not significantly different from those infants with bilateral mild VM.
In severe VM, neurodevelopmental outcomes are poor in most cases, in particular if there is progressive enlargement of the ventricles in utero. However, normal or near-normal development is reported and observed.
This underlines the urgent need for studies looking at the impact of a given neurodevelopmental delay on the overall quality of life at different time intervals, in order to estimate the actual burden of neurologic disabilities of children with VM.
Imaging Technique and Findings
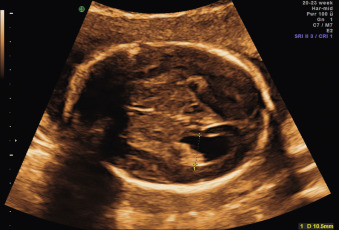
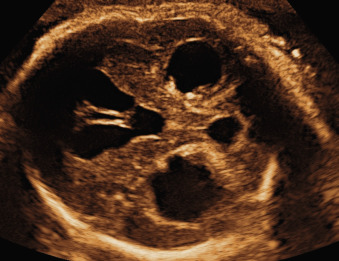
Ultrasound.
Measuring the width of the atria of the lateral cerebral ventricles is recommended as part of the routine anomaly scan. The lateral ventricle should be measured in the axial plane at the level of the cavum septi pellucidi, with the calipers aligned with the internal borders of the medial and lateral walls of the ventricle, and at the level of the glomus of the choroid plexus (the enlarged portion at the junction of the body of the lateral ventricle with the occipital and temporal horns of the lateral ventricles), opposite to the parietooccipital fissure ( Fig. 43.6 ). The diameters of both atria can often be measured on a coronal plane if shadowing of the proximal skull bones can be avoided; both choroid plexus can be seen, and measurements perpendicular to the long axis of the ventricles can be done, positioning calipers as before. This approach allows comparison of both lateral ventricles; it is also in close agreement with MRI measurements ( Figs. 43.7 and 43.8 ).