 Figure 13.1 A,B: In his monograph, Die Mischgeschwulste der Niere (the mixed tumor of the kidney), published in Leipzig in 1899, Max Wilms described, as his first illustrative case, “Niven Tumor von einem 3 jahrien Mädchen” (“renal tumor of a 3-year-old girl”). C: Wilms described the blastemal, epithelial (tubules), and stromal elements seen on microscopic examination of “mixed” renal tumor. |
the short arm of chromosome 11 (11p15). The putative second Wilms tumor suppressor gene, located at this site, is called WT2. WT2(LP15) has effects on IGF2, the H19 tumor suppressor gene, and the P57 cell cycle regulator (25, 26, 27, 28).
Table 13.1 Analysis for the Joint Effect of LOH at 1p and 16q for (A) Stage I/II and (B) Stage III/IV Favorable Histology Patients (29) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
seen in Wilms. The tumor consists of spindle-shaped cells in interlacing bundles adjacent to renal parenchyma where there are foci of cystic or dysplastic tubules. The treatment of choice is nephrectomy. Local recurrence is unusual. Nonetheless, adequate margins of resection should be obtained, although recurrence even after operative rupture or positive margins at resection is rare. Distant metastases are also rare. The actuarial 2-year survival rate is excellent at 98% (34, 35, 36, 37).
Table 13.2 Classification of Pediatric Renal Tumors | |||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||
Table 13.3 Revised International Society of Pediatric Oncology Working Classification of Renal Tumors of Childhood (2001) for Pretreated Cases | ||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||
Table 13.4 Children’s Oncology Group Classification of Renal Tumors | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
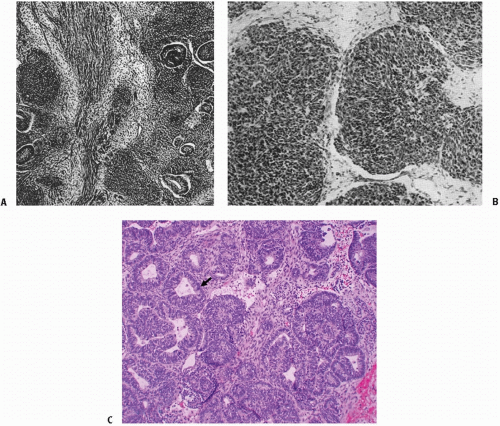 Figure 13.2 A,B: James B. Ewing, renowned for the four editions of his book Pathology of Neoplasia (Philadelphia, PA: WB Saunders; 1942) and his identification for the tumor now called Ewing sarcoma, also provided excellent histologic descriptions of Wilms tumor. These slides, from Ewing’s book, show the topography of Wilms embryonal tumor of the kidney with (A) epithelial tubules lining in masses of spindle and polyhedral cells and (B) the small round cells of the embryonal tumor infiltrating adjacent tissue. C: Favorable histology triphasic Wilms tumor with predominantly epithelial (tubular) differentiation (arrow). |
the kidney, which might affect the likelihood of complete resection. In a recently revised definition, focal anaplasia refers to anaplasia that is sharply localized in the primary tumor, without significant nuclear or mitotic atypia in the remainder of the lesion. Diffuse anaplasia is either nonlocalized anaplasia, localized anaplasia with severe nuclear unrest elsewhere in the tumor, anaplasia outside the tumor capsule or in metastases, or anaplasia found in a random biopsy taken from the tumor.
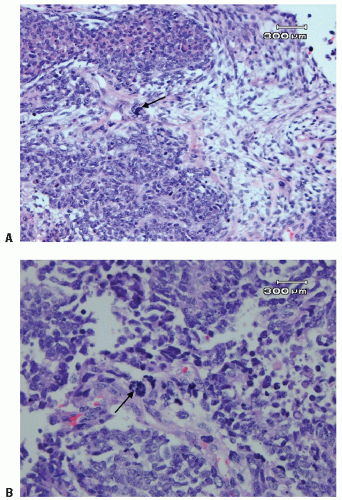 Figure 13.3 Anaplastic Wilms tumor with a large dark hyperchromatic nucleus (arrow) (A) and multipolar mitosis (arrow) (B). |
children entered in the study. Response of CCSK to therapy was poor until adriamycin and local irradiation were added to the treatment program. The 4-year relapse-free survival rate for patients with stages I-IV CCSK treated with vincristine, adriamycin, and actinomycin D on NWTS-3 was 71% (39).
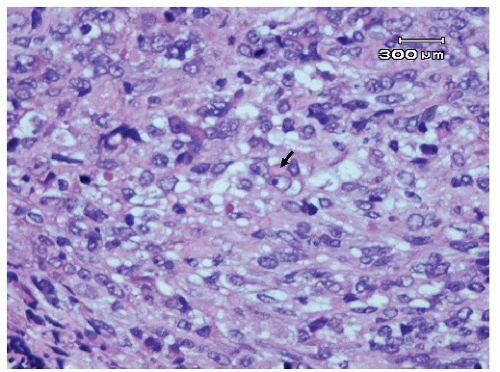 Figure 13.4 Rhabdoid tumor (RTK) with tumor cells with large eccentric vesicular nuclei, prominent nucleoli, and cytoplasmic inclusions (arrow). |
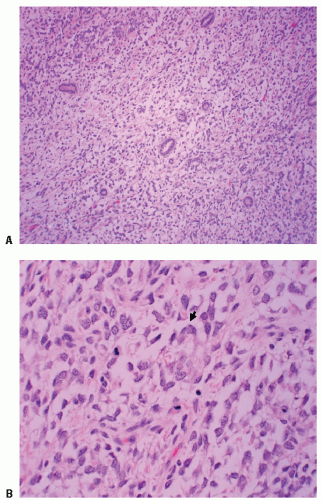 Figure 13.5 Clear cell sarcoma (CCSK) with tumor cells composed of uniform clear cytoplasm surrounding entrapped renal tubules: (A) low power, (B) high power showing prominent vacuolated cytoplasm (arrow). |
bilateral Wilms tumor. Others believe that imaging is useful but not definitive and that a surgical exploration of the contralateral kidney remains essential during the surgical approach to the primary tumor (17,47).
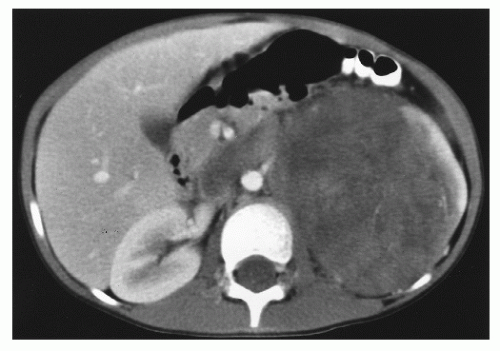 Figure 13.6 A 5-year-old girl presented with intermittent sharp abdominal pain, nausea, vomiting, weight loss, and hematuria. Abdominal computed tomography showed a 7.9- to 12.5-cm mass at the left renal region, beginning immediately subdiaphragmatically and extending to just below the aortic bifurcation. There was a calcified rim superiorly and a heterogeneous parenchymal enhancement. Left renal vein invasion and inferior vena cava thrombosis were observed. On exploratory laparotomy, tumor was palpated in the inferior vena cava. The tumor was resected, with some tumor spillage from a weak point on Gerota fascia. Tumor thrombus was removed from the inferior vena cava. Pathology showed a favorable histology Wilms tumor, 13.5 cm in greatest diameter, invading through the renal capsule to the inked surgical margin. Rupture of the renal capsule was identified. Tumor was present at the renal vein margin of the tumor thrombosis with focal invasion of the vein wall. No lymph nodes were involved. For pathologic stage III Wilms tumor, favorable histology, the child was treated with chemotherapy and flank irradiation. There was no evidence of persistent or recurrent tumor 3 years after diagnosis. |
Table 13.5 Children’s Oncology Group Staging of Wilms Tumor | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
|
posttreatment histology in these two SIOP studies and in 83 patients who underwent prenephrectomy chemotherapy in NWTS-3 showed no evidence that prenephrectomy therapy altered the detection of anaplastic histology (62, 63, 64). It appears that the SIOP presurgical therapy approach does not influence histology.
Table 13.6 Staging System Used in SIOP Wilms Tumor Trial 1 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
|
was discerned in the 10- to 40-Gy range, delays of up to 10 days in initiating postoperative irradiation appeared acceptable, and whole- abdomen irradiation was not found to be necessary for tumor spills confined to the flank or for prior tumor biopsy (17,41,57). In these patients, limited radio therapy fields sufficed.
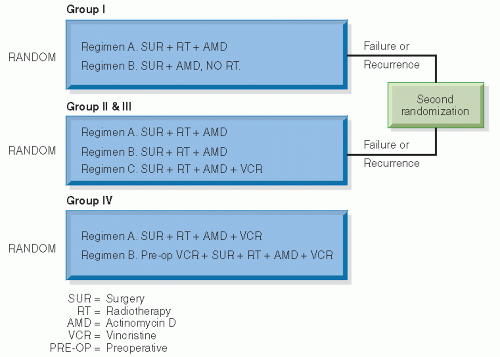 Figure 13.7 The design of NWTS-1. (From D’Angio GJ. National Wilms’ Tumor Study. Seattle, WA: NWTS Data and Statistical Center; 1991 [Informational Bulletin #19], with permission.) |
Table 13.7 NWTS-1 Results | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
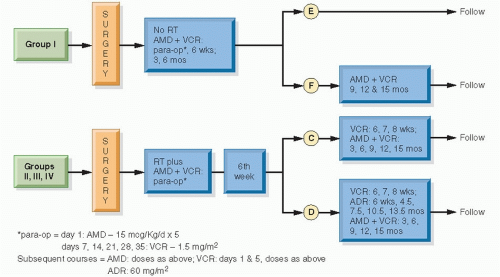 Figure 13.8 The design of NWTS-2. (From D’Angio GJ, Tefft M, Breslow N, et al. Radiation therapy of Wilms’ tumor: results according to dose, field, post-operative timing and histology. Int J Radiat Oncol Biol Phys. 1978;4:769-780, with permission.) |
Table 13.8 NWTS-2 Results | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
adriamycin clearly beneficial and therefore necessary in stages II and III FH? Will the addition of cyclophosphamide improve survival in stages I-III UH and in stage IV FH and UH?
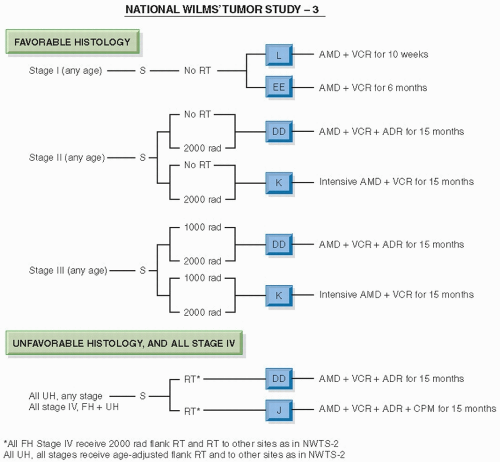 Figure 13.9 The design of NWTS-3. (From D’Angio GJ, Evans A, Breslow N, et al. The treatment of Wilms’ tumor: results of the Second National Wilms’ Tumor Study. Cancer. 1981;47:2302-2311, with permission.) |
adriamycin, compared with pulsed, intensive actinomycin D, vincristine, and adriamycin; all patients received radiotherapy (for FH, 10.8 Gy to the abdomen; 12 Gy WLI where appropriate; for stages II-IV anaplastic tumors, a sliding scale of radiation dosage was used, as in NWTS-1). Stages II-IV anaplastic Wilms tumors were treated with appropriate irradiation followed by actinomycin D, vincristine, and adriamycin, compared with these three drugs plus cyclophosphamide. Stages II-IV FH and stages I-IV CCSK were treated with 26 or 54 weeks of chemotherapy (Fig. 13.10).
Table 13.9 NWTS-3 Results | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Table 13.10 NWTS-3 Relapse Rates | ||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||
treatment groups (77,78). Comparing the results from NWTS-4 with those of the UKCCSG Wilms Tumor Study 2, reported contemporaneously, we see surprisingly similar results (Table 13.12) (79). The risk of local recurrence of patients enrolled in NWTS-4 is shown in Table 13.13.
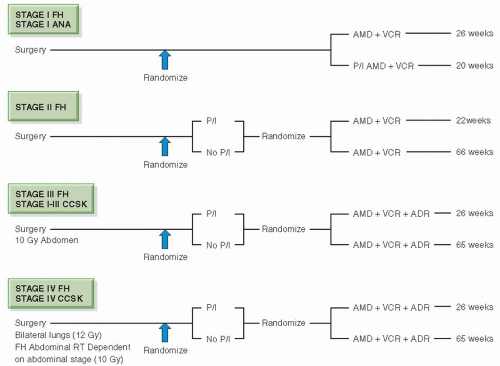 Figure 13.10 NWTS-4 simplified schema. Stage IV anaplastic tumors continued the randomization of NWTS-3. (From Thomas PRM. Wilms’ tumor: changing role of radiation therapy. Semin Radiat Oncol. 1997;7:204-211, with permission.) |
10.5 Gy in 50 patients. The overall abdominal and systemic tumor control rates were 97% and 93%, respectively. The detection of peritoneal implants was not associated with inferior survival after this therapy. The 5-year relapse-free survival rates with and without peritoneal implants were 90% and 83%, respectively (81).
Table 13.11 NWTS-4 Results | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


