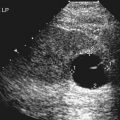
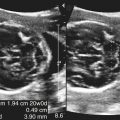
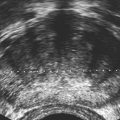
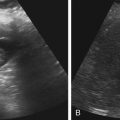
• Describe anomalies that may be identified with prenatal sonography. • Identify the sonographic features that differentiate fetal anomalies. • List anomalies that may benefit from in utero treatment. • Identify anomalies that are incompatible with survival after birth. • List other methods of fetal evaluation used in the diagnosis of anomalies. CLINICAL SCENARIO A young woman is seen for a sonogram for late prenatal care. The sonogram reveals a single, live fetus at 34 weeks’ gestation. In addition to the findings in Figure 25-1, a single umbilical artery and an absent stomach bubble are noted. What is the most likely diagnosis and prognosis? Patients may be referred for an obstetric sonogram for a suspected fetal anomaly. With little additional information, the sonographer must survey the fetus carefully to identify and clarify the nature of the abnormality. Sonographers must be familiar with a variety of fetal anomalies. Proper documentation and subsequent diagnosis must be made for a meaningful discussion with the patient of pregnancy, treatment, and delivery options. This chapter presents many of the fetal anomalies that are identified with sonography (Table 25-1). Other chapters review renal anomalies (see Chapter 20), neural tube and abdominal wall anomalies (see Chapter 23), chromosomal anomalies (see Chapter 24), and fetal heart defects (see Chapter 26). TABLE 25-1 Hydrocephalus is defined as a dilated ventricle with enlargement of the head, and ventriculomegaly is dilation of the ventricles without enlargement of the head, although these terms are sometimes used interchangeably. Hydrocephalus occurs in 0.5:1000 to 3:1000 live births and is associated with high morbidity and mortality rates.1 Hydrocephalus may be the result of a congenital anomaly or acquired; when hydrocephalus leads to intracranial pressure, brain parenchyma may also be compromised. In addition, ventricular enlargement may result from compensation for atrophy or abnormal development of brain parenchyma. The outcome is variable depending on the associated anomalies and the severity of the ventricular enlargement. The diagnosis of ventriculomegaly can be made when the atrium of the lateral ventricle exceeds 10 mm (Fig. 25-2) in the second and third trimesters of pregnancy. When ventriculomegaly is identified, an evaluation of all fetal measurements can help to determine the degree of ventriculomegaly and whether fetal head enlargement is present. A thorough sonographic evaluation should also be performed in a search for associated anomalies. Holoprosencephaly encompasses a range of severity characterized by incomplete or lack of cleavage of the forebrain. The condition has been identified in 1:10,000 to 1:20,000 births,2 and many affected fetuses spontaneously abort in the first trimester. The most severe form, alobar holoprosencephaly, manifests with a complete failure of separation of the forebrain with subsequent fusion of the thalami, a single ventricle, and fusion of the cerebral hemispheres. Absence of the falx, corpus callosum, olfactory bulbs, and optic tracts is also noted. Semilobar holoprosencephaly is seen with partial cleavage of the forebrain and absence of the corpus callosum and olfactory bulbs. Identification of the mildest form, lobar holoprosencephaly, may be difficult because separation of the cerebrum is evident, but fusion of the lateral ventricles and absence of the corpus callosum may be noted. The outcome is variable depending on the severity, but the most severe forms of holoprosencephaly carry an extremely poor prognosis. Holoprosencephaly is associated with severe facial anomalies, including cyclopia, ethmocephaly, cebocephaly, and facial clefts. Holoprosencephaly is also associated with multiple chromosomal anomalies, most commonly trisomy 13. In addition, holoprosencephaly may be identified in many syndromes, including Edwards’ syndrome (trisomy 18), Smith-Lemli-Opitz syndrome, and Pallister-Hall syndrome.3 Holoprosencephaly may also be transmitted through autosomal dominant, autosomal recessive, and X-linked modes. There is a 2:1 female-to-male prevalence and a 13% risk of recurrence.2 Also, several teratogenic effects have been linked to holoprosencephaly, including hyperglycemia (in diabetes) and maternal infections. Sonographic identification of holoprosencephaly is easier in its most severe form, and identification of a single ventricle (Fig. 25-3, A) surrounded by brain parenchyma is diagnostic. The thalami appear fused, and the falx is absent, as are the cavum septi pellucidi and corpus callosum. In semilobar holoprosencephaly (Fig. 25-3, B and C), variable fusion may be identified, and lobar holoprosencephaly may be difficult to distinguish from ACC, which should be suspected when the cavum septi pellucidi is not identified. When enlargement of the ventricle is identified, holoprosencephaly may be difficult to differentiate from hydranencephalus or severe hydrocephalus, unless the characteristic facial anomalies are also identified. When holoprosencephaly is identified, the face should be meticulously surveyed for the presence of associated anomalies (Fig. 25-3, C-G), including facial clefts, hypotelorism, a nose with a single nostril, a proboscis, and cyclopia. Other anomalies, such as a heart defect, polydactyly, or encephalocele, may suggest association with a chromosomal anomaly or syndrome. Hydranencephaly is characterized by destruction of the brain parenchyma with replacement by cerebrospinal fluid. It may result from vascular compromise leading to occlusion of the internal carotid artery4; however, other causes including congenital infections and cocaine abuse have been linked. The destruction to the brain usually occurs in the second trimester of pregnancy and may be preceded by a normal sonographic evaluation. Hydranencephaly is a rare anomaly with a grave outcome. The sonographic findings of hydranencephaly (Fig. 25-4) include liquefaction of the brain parenchyma with replacement by cerebrospinal fluid. The brainstem is intact and visible on sonographic evaluation. The head may be normal or large in size. Hydranencephaly can be differentiated from severe hydrocephaly by a lack of surrounding brain parenchyma. DWM is characterized by absence or dysplasia of the cerebellar vermis and maldevelopment of the fourth ventricle with replacement by a posterior fossa cyst. It occurs in 1:30,000 births5 and is frequently associated with ventriculomegaly. A less severe form, referred to as the Dandy-Walker variant, may occur with dysplasia of the cerebellar vermis without enlargement of the posterior fossa. DWM may be isolated or associated with syndromes, aneuploidy, and maternal infections.6 DWM also has been linked to other intracranial anomalies, including ACC. Of infants who survive, 40% to 70% are intellectually impaired.5 The diagnosis of DWM can be made with the identification of a posterior fossa cyst, with a resultant enlarged cisterna magna and splaying of the cerebellar hemispheres (Fig. 25-5) from the abnormal development of the cerebellar vermis. Hydrocephalus (see Fig. 25-2, A) frequently accompanies DWM. A thorough search for additional anomalies may assist in determination of whether DWM is isolated or associated with a chromosomal anomaly or syndrome. ACC is considered relatively common occurring in 0.3% to 0.7% of the general population and affecting 2% to 3% of individuals with mental retardation,7 although the condition may go undiagnosed. The corpus callosum connects the cerebral hemispheres and aids in learning and memory. ACC may be found in isolation but is often associated with other anomalies of the central nervous system, a variety of chromosomal anomalies, syndromes, and metabolic diseases.7 The outcome is variable depending on the association and severity of other anomalies, but ACC can be asymptomatic or have a grave prognosis when severe anomalies are identified. The diagnosis of ACC should be suspected when the cavum septi pellucidi is absent (Fig. 25-6) because the corpus callosum develops with the cavum septi pellucidi. In addition, colpocephaly, defined as mild ventriculomegaly with dilation of the occipital horns, giving the ventricle a teardrop appearance, may be identified. The lateral ventricles are also displaced laterally, and the third ventricle may appear dilated and displaced superiorly. The sonographic examination should include a thorough search for additional anomalies that would suggest a more extensive central nervous system anomaly, aneuploidy, or syndrome. Cleft lip or cleft palate is characterized by a defect in the upper lip or palate in the roof of the mouth. These anomalies constitute the most common congenital defects of the face, with occurrence of 1:700 to 1:1000.8 Cleft palate occurs in approximately 80% of cases of cleft lip8 and can be difficult to detect sonographically, especially in isolation. Cleft lip may be unilateral, bilateral, and median in location (Fig. 25-7). Unilateral clefts more commonly are left-sided. Median clefts and bilateral clefts frequently have an associated cleft palate with an increased frequency of associated anomalies, including aneuploidy, skeletal anomalies, and syndromes. Micrognathia is defined as a small or recessed chin. Micrognathia has been associated with aneuploidy, skeletal anomalies, and multiple syndromes. The most common anomaly associated with micrognathia is trisomy 18.9 The outcome for fetuses with micrognathia depends on the associated anomalies. Congenital diaphragmatic hernia (CDH) is a herniation of abdominal contents into the thorax through a defect in the diaphragm; it has an incidence of 3:10,000 to 5:10,000 births.10 The cause is unknown, although genetic and teratogenic factors have been associated. This uncommon malformation is most commonly left-sided and is associated with a high morbidity and mortality rate with a survival rate of 60%.10 Although associated anomalies influence the outcome, isolated defects also are associated with significant mortality secondary to respiratory complications from pulmonary hypoplasia. Because left-sided defects occur more frequently, the most common sonographic finding is identification of the stomach above the diaphragm (Fig. 25-9). The intestines may also be identified in the thorax, and the heart is malpositioned in the chest. Identification of the absence of an intraabdominal stomach is important to differentiate between CDH and cystic lung defects. Right-sided defects may show the liver and gallbladder within the thoracic cavity. Polyhydramnios and hydrops may also be identified in association with CDH. A thorough search for additional anomalies that may affect pregnancy management is imperative. Congenital cystic adenomatoid malformation (CCAM) is a rare lung abnormality characterized by an overgrowth of terminal bronchopulmonary tissue occurring in 1:25,000 to 1:35,000 pregnancies.11 Five classifications have been defined, based on pathologic findings and cyst size. Type 0 is described as an acinar dysplasia. Types I and II are macrocystic masses that consist of multiple large cysts (type I) or a single large cyst and smaller cysts less than 1 cm (type II). Type III is a microcystic variety that appears as a large echogenic mass. Type IV is characterized as a peripheral cyst.11 CCAM is usually unilateral, involving one lobe of the lung. There is no gender prevalence. CCAM may be associated with hydrops, polyhydramnios, and pulmonary hypoplasia, all of which are associated with a poor outcome. Some lesions identified in the prenatal period may involute spontaneously, although they may become symptomatic after birth from infection or hemorrhage; malignant transformation has also been reported. Early diagnosis is important for management of pregnancy and delivery. Delivery at a hospital with a neonatal intensive care unit should be arranged because newborns may require respiratory support or surgery or both. Asymptomatic infants should have a chest radiograph, even if the CCAM appears to regress in the perinatal period, and evaluation with computed tomography (CT) should be considered in light of the fact that a normal chest radiograph may be seen in infants with an abnormal CT.12 The sonographic appearance of CCAM type I is one or more large cysts in the thorax, type II appears as multiple small cysts, and type III (Fig. 25-10) appears as a large echogenic mass. Displacement of the fetal heart, polyhydramnios, and hydrops may also be identified. Sonography may be used to monitor for possible regression of the lesion.
Fetal Anomaly
Diagnosis
Sonographic Findings
Hydrocephaly
Enlarged head; atrium of lateral ventricle >10 mm
Holoprosencephaly
Single ventricle, fused thalami, absent cavum septi pellucidi and corpus callosum, absent falx; facial anomalies (clefts, hypotelorism, cyclopia, proboscis)
Hydranencephaly
Liquefaction of brain parenchyma; brainstem may be identified
DWM
Posterior fossa cyst with enlarged cisterna magna, splaying of cerebellar hemispheres with absent or dysplastic vermis; commonly associated with hydrocephalus
ACC
Absent cavum septi pellucidi, mild ventriculomegaly with dilated occipital horns; lateral displacement of lateral ventricles and superior displacement of third ventricle; dilated third ventricle
Cleft lip
Disruption of soft tissue of upper lip; bilateral cleft may appear to have premaxillary mass
Micrognathia
Recessed chin
CDH
Abdominal contents, usually in stomach, in thorax; malposition of fetal heart; absence of intraabdominal stomach
CCAM
Type I, multiple large cysts; type II, visible cysts <1 cm; type III, large echogenic mass in thorax
Pulmonary sequestration
Echogenic mass, may be triangular; separate blood supply from aorta
PE
Fluid collection surrounding lungs; unilateral or bilateral
Duodenal atresia
Dilated stomach and proximal duodenum appearing as double bubble, polyhydramnios common
Bowel obstruction
Dilated loops of bowel proximal to obstruction
Meconium peritonitis
Calcifications on peritoneal surfaces; meconium pseudocyst; associated ascites or polyhydramnios may be identified
Ascites
Collection of fluid in fetal abdomen outlining organs and bowel
Abdominal cysts
Usually round, thin-walled anechoic structure; location and gender should be noted to identify origin
SCT
Heterogeneous or complex mass extending from fetal sacrum
Thanatophoric dysplasia
Significant micromelia, narrow thorax with protuberant abdomen, bowed limbs, macrocephaly with frontal bossing, trilobular skull may be identified
Achondrogenesis
Extreme micromelia, variable degrees of ossification of spine and calvaria, narrow thorax and ribs, rib fractures may be noted
Achondroplasia
Rhizomelia, trident hand, macrocephalus with frontal bossing, depressed nasal bridge
SRPS
Micromelia, narrow thorax, short ribs, polydactyly, cleft lip and palate may be identified
OI
Decreased ossification of bones, compressible calvaria, narrow ribs, short limbs, multiple fractures
Fetal Brain
Hydrocephaly
Sonographic Findings
Holoprosencephaly
Sonographic Findings
Hydranencephaly
Sonographic Findings
Dandy-Walker Malformation
Sonographic Findings
Agenesis of the Corpus Callosum
Sonographic Findings
Fetal Face
Cleft Lip
Micrognathia
Fetal Thorax
Congenital Diaphragmatic Hernia
Sonographic Findings
Congenital Cystic Adenomatoid Malformation
Sonographic Findings
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Fetal Anomaly