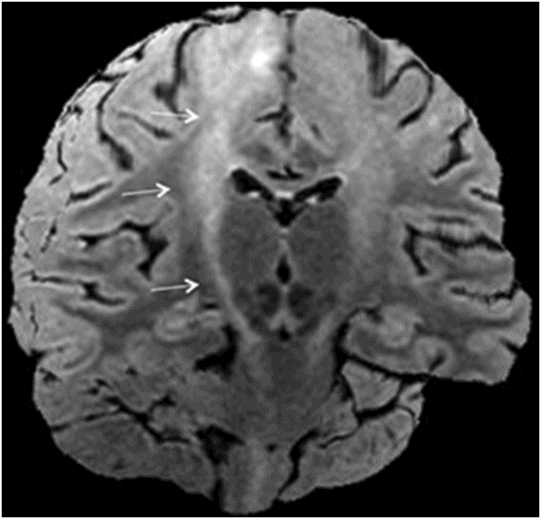
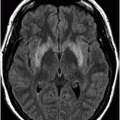
Axial FLAIR image of the brain at the level of motor cortex.
Primary Lateral Sclerosis
Primary Diagnosis
Primary lateral sclerosis Mills variety
Differential Diagnoses
Amyotrophic lateral sclerosis
Uncomplicated forms of hereditary spastic paraplegia
Multiple sclerosis
Adrenoleukodystrophy and metachromatic leukodystrophy
Vitamin B12 deficiency
HTLV-1 and HIV infection
Compressive cervical myelopathy
Imaging Findings
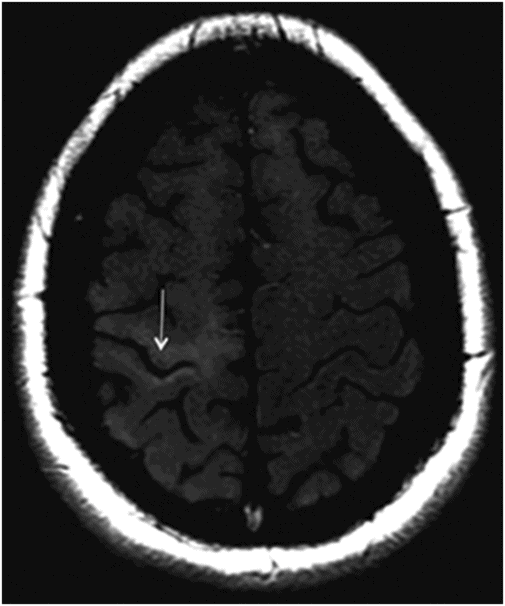
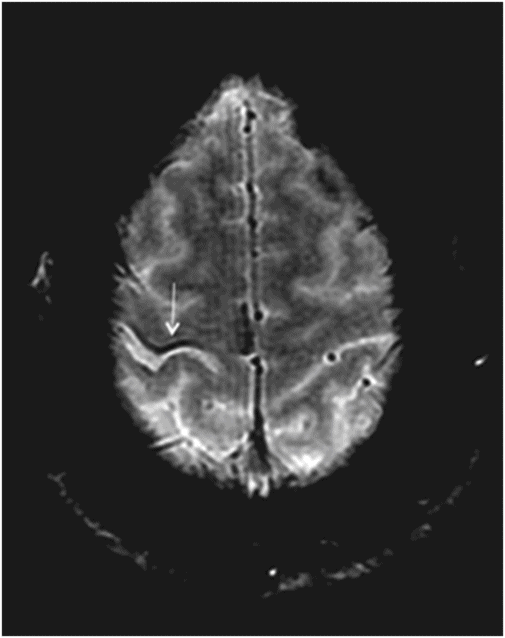
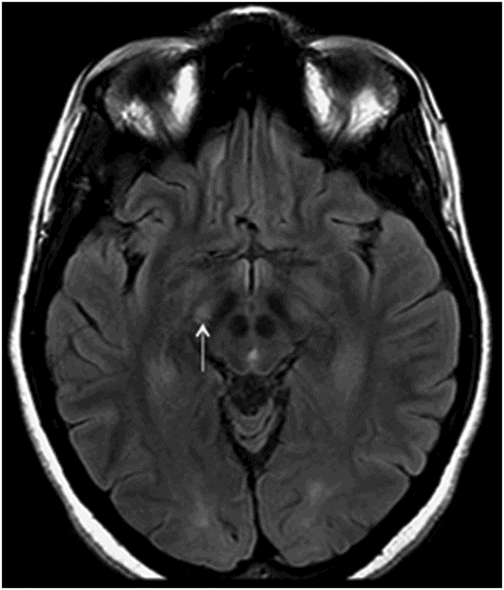
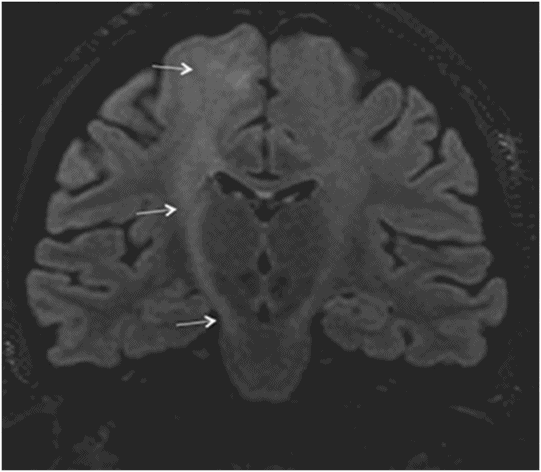
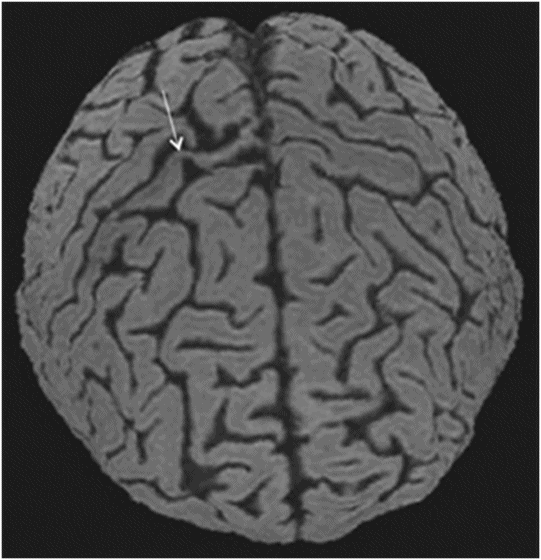
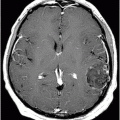
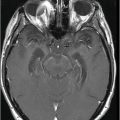
Fig. 14.1: Axial FLAIR image demonstrated abnormal hyperintense signal with focal cortical thinning and volume loss in the right motor cortex (arrow). Fig. 14.2: T-MTC (magnetization transfer contrast) sequence image is more sensitive than conventional FLAIR sequence in depicting the signal changes. Fig. 14.3: Axial GRE image showed T2 shortening in the motor cortex (arrow) representing iron deposition. Fig. 14.4: Axial FLAIR image showed asymmetric hyperintense signal in the right cerebral peduncle (arrow). Fig. 14.5: Coronal FLAIR volumetric 3D reconstruction sequence and Fig. 14.6: Oblique 3D reconstruction image also denoted abnormal signal changes (arrows) along the corticospinal tract. Fig. 14.7: Superior view of the surface rendering image of the brain obtained from 3D volumetric FLAIR image showed the atrophied right motor cortex (arrow).
Discussion
On imaging, amyotrophic lateral sclerosis (ALS) has similar features to primary lateral sclerosis (PLS). The most distinctive PLS feature, however, is the lack of lower motor neuron involvement. On the contrary, ALS presents with upper and lower motor neuron involvement. Hereditary spastic paraplegia can have overlapping features but presents in a younger age group. It has various forms on inheritance with multiple pheno- and genotypes. Few multiple sclerosis (MS) cases present with diffuse thinning of corpus callosum, white matter changes, and volume loss. Clinically, the primary progressive and pure motor variant of MS can have a similar clinical presentation. However, the typical MRI demyelinating pattern involving the periventricular-pericallosal regions, callososeptal interface, brainstem, and spinal cord features helps in differentiating MS and its variants from PLS.
Metachromatic leukodystrophy and adrenoleukodystrophy have multiple types and their adult forms tend to present earlier in life. The distribution pattern is symmetric with periventricular and central deep white matter changes and is associated with cognitive disturbances as the disease progresses. HTLV-1 and HIV infections can have similar clinical presentation and HTLV-1 can have imaging appearances similar to PLS and ALS; however, serologic findings can exclude these infections as possible diagnoses. Vitamin B12 deficiency can often mimic the presentation of PLS but has predominant cord signal changes with posterior column involvement and symptoms. Apart from vitamin B12 deficiency, cervical compressive myelopathy is another treatable condition, which can manifest with a similar clinical presentation to PLS. Affected individuals present with intractable pain; cord compression is often evident on cervical spine imaging and lacks the brain imaging features of PLS.
Primary lateral sclerosis is a rare form of motor neuron disease, which is typically differentiated from the more common amyotrophic lateral sclerosis by the absence of lower motor neuron involvement. It is characterized by spinobulbar spasticity due to upper motor neuron degeneration. In 1992, Pringle et al. described eight cases and proposed the current clinical diagnostic criteria. Primary lateral sclerosis is a disease of middle age, occurring most frequently during the fourth to sixth decades of life. It is slowly progressive and is common in both sexes. The most common PLS manifestation is leg weakness and spasticity, followed by spastic dysarthria. Although upper extremity symptoms develop over time, they are uncommon at presentation. Symptoms may unilaterally affect one side of the body, before progressing to involve the other side. Bulbar symptoms initially manifest as dysarthria, followed by dysphagia, and may evolve into emotional lability and inappropriate laughing or crying, the so-called pseudobulbar affect. The diagnostic criteria involves adult onset, negative family history, gradual progression, spasticity for more than three years, and dysfunction clinically limited to the corticospinal tracts. Hematologic and biochemical investigations are normal but help in excluding pertinent conditions such as vitamin B12 deficiency, HIV, HTLV-1, syphilis, Lyme disease, and some leukodystrophies that can closely resemble PLS.
Pathologically, primary lateral sclerosis is limited to the primary motor cortex, corticobulbar, and corticospinal tracts. Although PLS lacks lower motor neuron involvement clinically, evidence of mild denervation with occasional fibrillation may be seen on electromyography. In addition, cortically evoked motor potentials are often absent in patients with PLS. When present, the central motor conduction time (CMCT) may be considerably prolonged, two to three times normal. There is no positive marker for PLS. Autopsy may help diagnosis by showing selective Betz cell loss in the central motor cortex.
Amyotrophic lateral sclerosis and PLS share MR imaging features including T2 hyperintensity within the corticospinal tracts and atrophy of the motor and premotor cortex. The atrophy may be noticed in the CT images. The typical pattern of hyperintensity in bilateral corticospinal tracts in the coronal images is also described as a wine glass appearance. A band of hypointensity involving the cortex of precentral gyrus in gradient T2* or susceptibility-weighted images are also described in the literature. Decreased fractional anisotropy is noted in the pyramidal tracts in diffusion tensor imaging. Criteria require disease duration of at least three years before a PLS diagnosis can be made: the lack of clinical involvement of lower motor neurons will help differentiate PLS from ALS.

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


