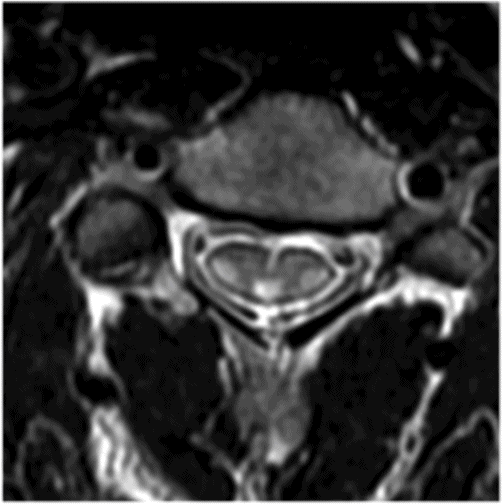
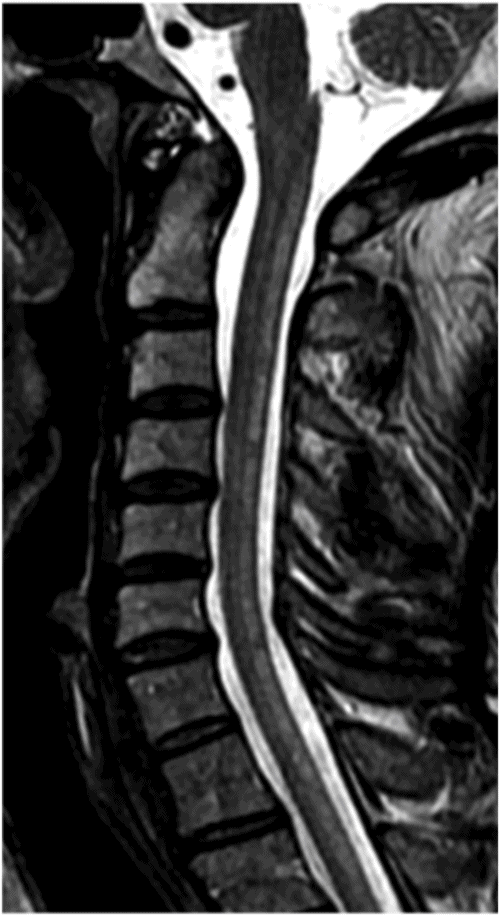
Axial T2WI through the midcervical cord.
Cerebrotendinous Xanthomatosis
Primary Diagnosis
Cerebrotendinous xanthomatosis
Differential Diagnosis
Refsum disease
Imaging Findings
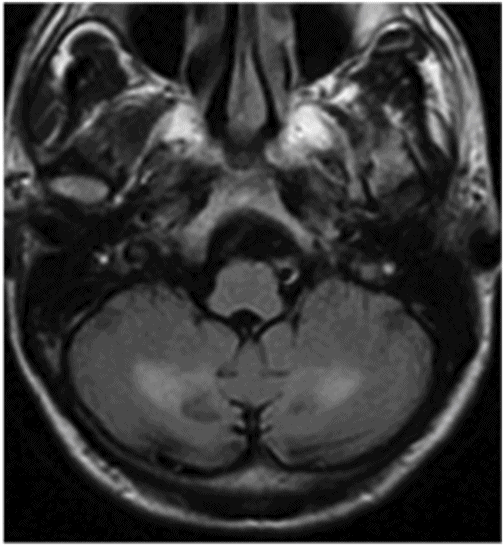
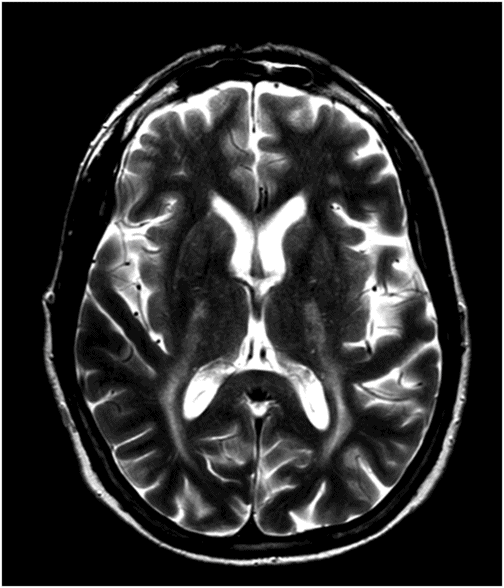
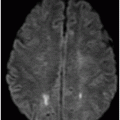
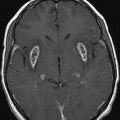
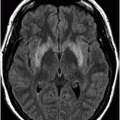
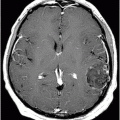
Fig. 16.1: Axial T2WI through the dentate nuclei demonstrated bilateral, symmetric T2 hyperintensity involving the dentate nuclei. Severe atrophy of the cerebellum was noted with prominent cerebellar sulci and secondary enlargement of the fourth ventricle. Subtle T2 hyperintensity in the superior aspect of the olives was also noted. Fig. 16.2: Axial FLAIR image through the cerebellar hemispheres demonstrated bilateral symmetric cerebellar white matter FLAIR abnormality. Fig. 16.3: Axial T2 image through the basal ganglia demonstrated bilateral, symmetric abnormal T2 signal in the periventricular white matter that extends posteriorly from the periatrial white matter to the globus pallidi through the posterior limb of the internal capsule. Fig. 16.4: Axial T2WI image through the midcervical spinal cord demonstrated bilateral, symmetric T2 hyperintensity restricted to the lateral and posterior columns. Fig. 16.5: Midsagittal T2WI through the cervical spine demonstrated longitudinal involvement of the posterior column.
Discussion
This patient has typical clinical presentation of cerebrotendinous xanthomatosis (CTX), including cerebellar symptoms, weakness, posterior column symptoms, bilateral muscular weakness, history of cataract surgery, and characteristic tendinous xanthomas in the prominent tendons. Bilateral, symmetric T2 abnormalities involving the dentate nuclei, periventricular white matter, internal capsule, globus pallidi, and lateral and posterior column are also consistent with the diagnosis.
Refsum disease typically presents with retinitis pigmentosa, peripheral neuropathy, cerebellar ataxia, and elevated CSF protein levels, without an increase in the number of cells. The typical age of presentation is late in the first decade through the third decade of life. Cataracts and CNS imaging findings are not suggestive of Refsum disease.
A rare autosomal recessive disorder, CTX is characterized by the accumulation of cholesterol and cholestanol, typically in the CNS and tendons. The mutation localizes in the CYP27 gene that encodes the mitochondrial 27-hydroxylase enzyme. Most patients have onset of symptoms in childhood and often present with chronic diarrhea and bilateral cataracts. Symptoms that are more specific usually manifest late in childhood, adolescence, or even in young adulthood, delaying diagnosis. Occurring in over 90% of patients with CTX, neurologic signs and symptoms generally manifest in the second and third decades of life and include cerebellar symptoms (ataxia, tachylalia, and dysarthria), pyramidal dysfunction, spastic paraparesis, tetraparesis, and polyneuropathy. Mental retardation, epilepsy, and psychiatric disorders may also occur. Tendon xanthomas typically affect the Achilles tendon, but also can occur in the tendon of the quadriceps muscle, the triceps muscle, and finger extensors. Clinical triad of CTX is cataract, tendon xanthomas, and progressive neurologic impairment; however, this classic triad of symptoms may not be present in all CTX patients.
The typical CTX imaging appearance, present in the majority of patients, consists of bilateral, symmetric T2 hyperintensity involving the dentate nuclei and adjacent cerebellar white matter. Other very characteristic but less common MRI signs are bilateral, symmetric T2 abnormality involving the globus pallidi and adjacent internal capsules, cerebral peduncles, and olives. These suggestive imaging findings are frequently accompanied by additional, non-specific findings such as focal or diffuse cerebral and cerebellar white matter T2 signal abnormality with varying degree of cerebral and cerebellar atrophy. On MR spectroscopy, low NAA peak, high lactate peak, and a prominent lipid peak can be seen with short TE spectra.
Laboratory findings include normal or slightly increased levels of serum cholesterol and a significant increase in urine cholestanol. Cerebrospinal fluid analysis may show increased protein levels and cholestanol.
Typically, the diagnosis is suggested by demonstrating abnormally high bile alcohol levels in urine. The confirmation can be done by validating the lack of 27-hydroxylase enzyme activity in cultured fibroblasts, liver, and leukocytes, and through DNA mutational analysis.
Microscopic examination of the dentate nucleus and the cerebellar white matter shows thinning with significant neuronal loss and demyelination cracks with cholesterol crystals, reactive astrocytosis, hemosiderin deposits, and calcifications. Large fat accumulation is apparent within mononuclear cells with a foamy cytoplasm, which generally accumulate around blood vessels.

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


