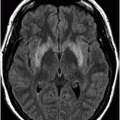
Axial FLAIR.
Huntington Disease
Primary Diagnosis
Huntington disease
Differential Diagnoses
Bilateral chronic caudate nuclei infarcts
Multiple system atrophy type P (putaminal)
Hypoxic ischemic injury
Imaging Findings
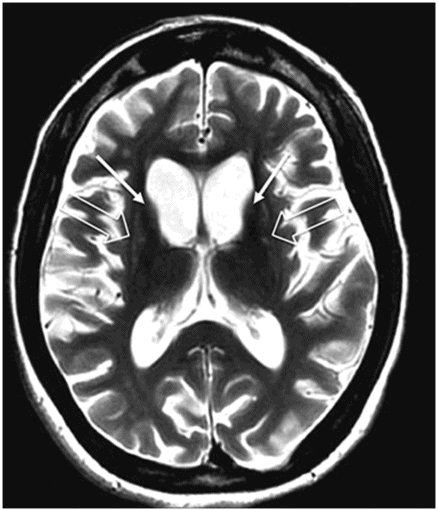
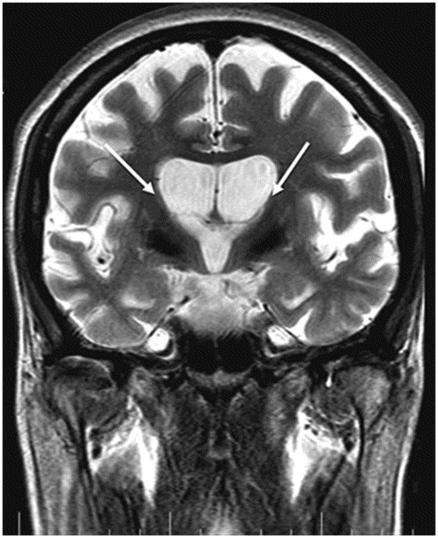
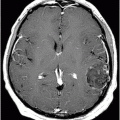
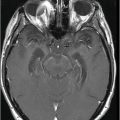
Fig. 17.1: Axial FLAIR and Fig. 17.2: Axial T2WI demonstrated diffuse brain atrophy as evidenced by enlargement of the subarachnoid spaces, particularly in the frontal regions (unexpected in a patient this age), and bilateral lateral ventricular enlargement, particularly the frontal horns, due to severe atrophy of the head of the caudate nuclei (thin arrows). Putaminal atrophy (large arrows) is also noted. Fig. 17.3: Coronal T2WI better showed compensatory enlargement of the frontal horns of the lateral ventricles (arrows).
Discussion
In a middle-aged patient, the progressive onset of a movement disorder and behavior changes in combination with imaging findings demonstrating bilateral caudate nucleus head and putaminal volume loss (striatal atrophy) is highly suggestive of a diagnosis of Huntington disease (HD).
Caudate infarcts are rare, representing approximately 1% of stroke (ischemic and hemorrhagic). Caudate nuclei infarction could result from involvement of any of the three arterial systems: 1) Heubner’s artery, a branch of the anterior cerebral artery, supplies the inferior portion of the head of the caudate and the anterior limb of the internal capsule; 2) anterior lenticulostriate arteries originating from the proximal part of the anterior cerebral artery, which supply the anterior portions of the caudate nuclei; and 3) the lateral lenticulostriate arteries from the middle cerebral artery, which supply the major portions of the caudate and the anterior internal capsule and putamen.
Although volume loss is an expected finding in chronic infarcts, bilateral symmetric caudate head infarct is very unlikely to be of vascular etiology. In addition, severe brain atrophy in this age is very suggestive of underlying neurodegenerative disease.
Multiple system atrophy type P (MSA-P) can cause putaminal volume loss, but not atrophy of the caudate nuclei. Additionally, patients with MSA-P are commonly in their sixth decade and present with Parkinsonism, bradykinesia, tremor, rigidity, and gait abnormalities. Owing to the discrepancy of imaging findings and clinical findings, the diagnosis of MSA-P is less likely.
Hypoxic ischemic injury (HII) is a known cause of bilateral basal ganglia volume loss, particularly in the chronic stage. It is also known that HII can involve not only the basal ganglia, but also the cerebral cortex and watershed areas of the brain. In adults, the most common causes of HII are cardiac arrest, drowning, suicidal attempt, or strangulation, which can induce a variable degree of neurologic abnormalities depending on the degree of injury. Owing to the absence of a consistent history, the possibility of HII is less likely.
Huntington disease is an autosomal dominant inherited neurodegenerative disorder that affects young adult patients with age ranging from 30 to 50 years. The disease is caused by a mutation in the HTT gene located in chromosome 4p16:3 that encodes the huntingtin protein. Although huntingtin is in multiple body tissues, it is most highly concentrated in the brain. Mutations to the huntingtin protein, implicated in neural transmission, protein transport, and apoptosis, lead to progressive neurodegeneration due to impairment of energy metabolism. Death usually occurs within one to two decades after the symptom onset.
The following clinical findings are suggestive of HD: progressive movement disorder (chorea); mental disturbances including cognitive decline, changes in personality and/or depression; and a positive family history. The natural history of patients with HD can be divided into three different stages. Initially, patients may experience clumsiness, agitation, irritability, apathy, anxiety, disinhibition, delusions, hallucinations, abnormal eye movements, and depression. In the following stage, chorea and involuntary movements become more prominent and voluntary activity, dysarthria, and dysphagia worsens. Later signs include rigidity, bradykinesia, severe chorea (less common), and an increase in the severity of motor disabilities. The affected individual, during the last stage, is often totally dependent, mute, and incontinent.
Neuropathologic features of HD primarily include a selective degeneration of GABAergic neurons in the caudate and putamen. The preferential degeneration of neurons of the indirect pathway of movement control in the basal ganglia provides the neurobiologic basis for chorea. Other regions of the brain that can be affected include the substantia nigra, hippocampus, and various regions of the cortex.
Any imaging modality suitable for structural brain evaluation is useful to support the clinical diagnosis of HD. Owing to its superior spatial and contrast resolution, MRI is the imaging modality of choice. The hallmark imaging feature of HD is caudate volume loss, which begins in the dorsal caudate and proceeds ventrally and laterally to encompass the putamen. Owing to caudate atrophy, there is enlargement of the frontal horns, giving them a box-like configuration. Caudate atrophy can be quantified using the frontal horn width-to-intercaudate distance ratio (FH/CC) as well as the intercaudate distance-to-inner table width ratio (CC/IT). Additionally, diffuse cortical atrophy is present, particularly in the frontal regions. Signal changes in the caudate and putamina may be noted, representing gliotic tissue.
Several studies using advanced MRI techniques have demonstrated brain changes in pre-HD patients, especially in white matter striatum and brain volumes. Magnetic resonance imaging can demonstrate significant striatal atrophy as many as 11 years prior to clinical onset of the disease.
Spectroscopy can show increased lactate peaks in the basal ganglia and in the occipital cortex of affected individuals. Markers of neuronal degeneration, decreased NAA/creatine and increased choline/creatine levels, can also be found. At present, there is no cure, nor any effective treatment to delay symptom onset or slow progression, and most current treatments aim to minimize symptoms.

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


