Frontotemporal Lobar Degeneration Associated with Fused in Sarcoma Protein
Primary Diagnosis
Frontotemporal lobar degeneration associated with fused in sarcoma protein
Differential Diagnoses
Frontotemporal lobar degeneration-tau
Frontotemporal lobar degeneration-TDP
Huntington disease
Neuroacanthocytosis
Imaging Findings
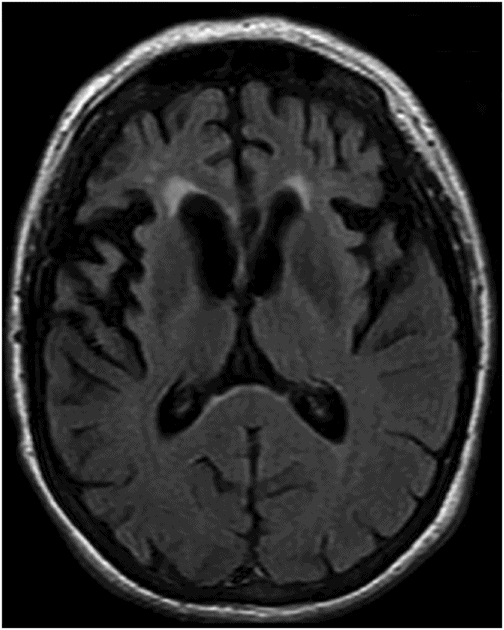
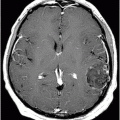
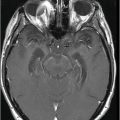
Fig. 18.1: Axial T2WI through the level of the basal ganglia demonstrated diffuse symmetric brain atrophy that is more severe in the bilateral frontal lobes. In addition, severe atrophy of the head of the caudate nuclei with unusual prominence of the frontal horns is noted. Fig. 18.2: Axial FLAIR sequence through the level of the basal ganglia inferior to Fig. 18.1 demonstrates diffuse symmetric brain atrophy that is more severe in the bilateral frontal and temporal lobes. In addition, the atrophy of the head of the caudate nuclei is noted.
Discussion
Frontotemporal lobar degeneration-tau (FTLD-tau) and frontotemporal lobar degeneration-TDP (FTLD-TDP) are the two most common pathologic findings in frontotemporal dementia (FTD); however, they are not associated with striatal atrophy. Huntington disease (HD), an autosomal dominant neurodegenerative disease, is characterized by severe caudate atrophy and a characteristic abnormal movement. As this patient did not have any known family history or chorea-like movement, HD was excluded. Lack of blood acanthocytes rules out neuroacanthocytosis, a heterogeneous group of genetically defined HD-like neurodegenerative diseases characterized by red blood cell acanthocytosis and chorea-like movements.
Frontotemporal dementia, a cluster of clinical syndromes resulting from the preferential neurodegeneration of the orbitomedial frontal lobe and temporal lobe in varying combinations, is extremely heterogeneous in terms of clinical presentation, histopathologic findings, imaging findings, and genetics. There are three well-known clinical syndromes categorized under FTD: 1) behavioral variant (bv-FTD), 2) progressive non-fluent aphasia (PNFA), and 3) semantic dementia (SD). Behavioral changes are the dominant clinical findings in bv-FTD, which preferentially affects the frontal lobe. The breakdown of spontaneous speech and difficulty understanding language are the key clinical findings in PNFA, which preferentially affects the perisylvian area of the dominant hemisphere, and fluent speech with prominent anomia, word finding difficulty, and difficulty in word recognition are the typical findings in SD due to preferential involvement of the dominant temporal lobe. Approximately 15% of patients with bv-FTD have associated motor neuron disease. This variant is recognized as a special subtype of FTD (FTD-MND) as it has a specific genetic linkage. In addition to these four clinical variants, progressive supranuclear palsy syndrome (PSPS) and corticobasal syndrome (CBS) are linked to a number of molecular pathologies targeting the frontal and temporal lobes that are collectively called frontotemporal lobar degeneration (FTLD). Parkinsonian features are noted in all FTD subtypes.
Unlike FTD, which is a clinical term, FTLD incorporates specific aspects of the pathologic and histopathologic findings. Similar to the three well-recognized clinical syndromes, FTLD can be subclassified: 1) FTLD-tau, characterized by tubulin-associated unit immunoreactive inclusions; 2) FTLD-TDP-43, characterized by transactive response DNA binding protein of 43 kDa immunoreactive inclusions; and 3) FTLD-FUS, characterized by fused in sarcoma immunoreactive inclusions. Both the TDP and FUS proteins are ubiquitinated and part of FTLD-U. Cases that are only positive for ubiquitin-immunoreactive inclusions are categorized as FTLD-UPS (UPS, i.e., ubiquitin proteasome system). Almost all of the FTD variants can be characterized by the three different FTLD patterns. Pick disease, corticobasal degeneration, and progressive supranuclear palsy (PSP) are pathologically categorized as FLTD-tau, as are other extremely rare histologic subtypes such as argyrophilic grains disease, sporadic multisystem tauopathy, and diffuse neurofibrillary tangle dementia with calcifications.
There is strong clinical association of each clinical subtype of FTD syndrome with all FTLD subtypes. Almost all cases of CBS and PSP are associated with FTLD-tau, whereas all of the FTD-MNDs are associated with FTLD-TDP. More than 80% of cases of SD are associated with FTLD-TDP, and up to 70% of PNFA cases are associated with FTLD-tau. Patients with bv-FTD are predominantly categorized as FTLD-tau or FTD-TDP. Young-onset bv-FTD is typically associated with FTLD-FUS.
Originally described in association with human cancers, FUS belongs to FET/TET family of multifunctional DNA/RNA binding proteins. Fused in sarcoma proteinopathies, linked to both amyotrophic lateral sclerosis (ALS) and FTLD, are characterized by the presence of inclusions that are immunoreactive for FUS in the cytoplasm and nuclei of both glial and neuronal cells. How FUS accumulation leads to neurodegeneration is still not well understood. FTLD-FUS clinically manifests as bv-FTD or FTD-MND characterized by severe progressive psychobehavioral alteration, negative family history, and striking striatal atrophy. FTLD-FUS encompasses approximately 10% of FTLD-U and approximately 3% of FTD syndrome. It has been shown that FTLD-FUS is associated with bilateral atrophy of the caudate nuclei, which can be used as an antemortem biomarker for FTLD-FUS subtype. In addition, FTLD-FUS is associated with bilateral symmetric extratemporal atrophy rather than the asymmetric atrophy of the frontal and temporal lobes in other subtypes of FTLD.

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


