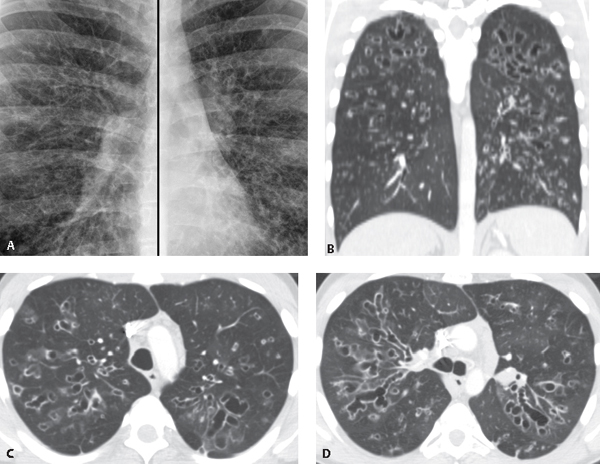
CASE 26 33-year-old man with wheezing, dyspnea and productive cough Coned-down composite PA chest radiograph (Fig. 26.1A) reveals extensive bilateral bronchiectasis manifesting as ring, tram track, nodular and reticular opacities most severe in the upper lungs. Unenhanced chest CT (lung window) coronal reformation (Fig. 26.1B) and axial images (Figs. 26.1C, 26.1D) demonstrate increased lung volumes (Fig. 26.1B) and extensive bronchiectasis in both lungs that predominantly involves the upper lobes; focal nodular and tubular opacities represent mucoid impaction (Figs. 26.1B, 26.1C, 26.1D). Note subtle parenchymal heterogeneity (mosaic attenuation) (Figs. 26.1B, 26.1C, 26.1D). Fig. 26.1 Cystic Fibrosis • Williams-Campbell Syndrome • Allergic Bronchopulmonary Aspergillosis (ABPA) • Other Causes of Bronchiectasis (e.g., chronic aspiration, healed tuberculosis) Cystic fibrosis is a hereditary, multi-system, genetically transmitted disease that affects exocrine tissues in the lung, pancreas, gastrointestinal tract, liver, salivary glands, and the male reproductive system. Cystic fibrosis is transmitted as an autosomal recessive trait. The responsible gene is located on the long arm of chromosome 7. The protein product of the cystic fibrosis transmembrane conductance regulator (CFTR) gene may undergo many different mutations that can lead to cystic fibrosis. Phenotypic variations occur in the magnitude of sweat chloride elevation, the presence and degree of pancreatic insufficiency, the age of onset, and the severity of pulmonary disease. In the lung, dysfunction of an amino acid protein impairs the ability of airway epithelial cells to secrete salt (and thus water), with resultant excessive reabsorption of salt and water. This leads to dessication of luminal secretions with mucous plugging. Mucociliary clearance decreases, predisposing to colonization by bacteria and recurrent infection.
 Clinical Presentation
Clinical Presentation
 Radiologic Findings
Radiologic Findings
 Diagnosis
Diagnosis
 Differential Diagnosis
Differential Diagnosis
 Discussion
Discussion
Background
Etiology
Clinical Findings
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


