32 Algodystrophy (Reflex Dystrophy, Complex Regional Pain Syndrome Type I)
The algodystrophy symptom complex describes a posttraumatic complication of multifactorial etiology that can lead to complete loss of function in an extremity. The patient’s history and clinical findings – in this case, especially the main symptom of pain – are the relevant diagnostic parameters. Therefore, imaging procedures are most useful in excluding concomitant diseases, as there are no specific morphological diagnostic criteria, the transition toposttraumatic changes being subtle. Three-phase scintigraphy can contribute to the diagnosis by identifying typical functional disturbances in the bone metabolism.
Pathogenesis and Clinical Symptoms
In the new nomenclature, the terms algodystrophy (synonyms:sympathetic reflex dystrophy, Sudeck disease) and causalgia, are subsumed under the generic term “complex regional pain syndrome” (CRPS). Type I (without morphologically identifiable nerve lesions) corresponds to algodystrophy, whereas type II (with defined nerve lesions) represents causalgia. CRPS covers a variety of symptoms with the diagnostic criteria listed in Table 32.1 .
The symptoms of type I and type II hardly differ. The degree of expression of these criteria depends on the stage of disease. Transitory osteopenia or transitory bone-marrow edema and “aggressive” regional osteopenia (e.g., of the humerus) also belong to the algodystrophic symptom complex.
The pathogenesis and pathophysiology of CRPS and accordingly also of algodystrophy, are not fully understood. Triggering events are usually injuries or surgery. The probability of CRPS manifestation increases with the severity of the trauma, but there is no direct correlation with the severity of CRPS. Neurologic, vegetative, vascular, humoral, and psychological factors play important roles. The disease has also been observed after injuries to the spinal chord, strokes, local infections, gout, heart attacks, pleural diseases, and thrombotic occlusions. Algodystrophy can also appear with neuromuscular diseases, diabetes, hypothyreosis, and metabolic disturbances, during pregnancy, during medical treatment (tuberculostatics, barbiturates, and antiepileptics), and can be manifested bilaterally. An important factor seems to be the involvement of the sympathetic nervous system with consecutive disturbance in perfusion and metabolism. Recent publications postulate involvement of the nonmyelinated, nociceptive C fibers, which possess both afferent and efferent neurosecretory functions. The released neuropeptides cause a neurogenic inflammation whose symptoms in experimentally induced disease resemble those of CRPS. Further investigations point to central-nervous disintegration. This assumption is supported by the observation that the regions affected by algodystrophy extend far beyond the borders of the original site of damage and can even migrate. The disease can also arise in the distribution region following injury to the spinal chord or a stroke.
The disease follows a prolonged course. It can be divided into three separate stages:
The initial stage is characterized by symptoms similar to those of inflammation, with the classic signs of pain, hyperthermia, redness, swelling, and limited range of articular motion. Sweat secretion is increased, and the hair and nails grow faster, as does the bone length among adolescents.
In the dystrophic stage, which is reached after about three months, circulation is diminished. Gray cyanosis appears with hypothermia, decreased sweat secretion, trophic disturbances with reduced growth of skin appendages, and reduced longitudinal bone growth. Pain at rest and during movement persists.
The stage of terminal atrophy is reached only after many months or even years. It is characterized by shiny skin resulting from atrophy of the corium layer, loss of muscle mass, and irreversible joint stiffness with contractures. The skin displays pale to blue cyanosis due to increased passive venous filling. The final result is a total loss of function in the affected extremity.


Diagnostic Imaging
The main importance of diagnostic imaging in algodystrophy is to rule out concomitant diseases (initially overlooked fractures, activated osteoarthritis, acute carpal tunnel syndrome, and osteomyelitis/arthritis), as well as to document the degree of bone demineralization induced by inactivity and its sequelae. Lesions assessed in diagnostic imaging are caused by disturbed perfusion, which leads to negative bone metabolism. None of the imaging procedures provides a particular characteristic that confirms the presence of algodystrophy.
We therefore emphasize here that the diagnosis of algodystrophy can only be made on the basis of clinical findings. Diagnosis of so-called Sudeck diseaseon the basis of a diffuse decalcification of the hand skeleton in radiographs alone is absolutely unacceptable.
Radiography
To obtain quantitatively reproducible radiographs suitable for follow-ups, the following points must be taken into account:
Use of high-definition screens (sensitivity < 100) or high-resolution digital imaging systems
Exposure quality that permits assessment of both bone structure and the thickness of the soft-tissue covering
Exposure of both the diseased hand and the healthy, contralateral hand on one survey radiograph in a dorsopalmar projection
Complete documentation of the imaging parameters (sensitivity class, focus size, kilovoltage, mAs, focus–film distance [FFD]).
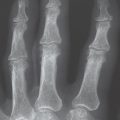
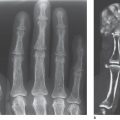
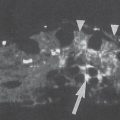
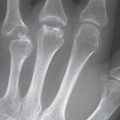
The radiograhic findings in algodystrophy are summarized according to stage in Table 32.2 and Figs. 32.1 and 32.2 .
Since skeletal changes do not appear until a few weeks after clinical symptoms have begun, a survey radiograph is not appropriate for early diagnosis. The pattern of resorption seen in stages II and III does not confirm the presence of algodystrophy because the transition to inactivity demineralization isindistinct.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


