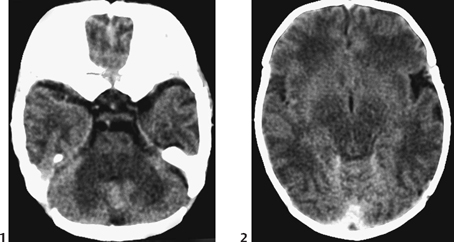
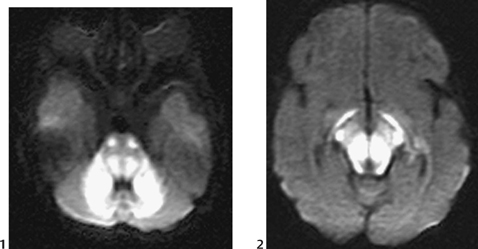
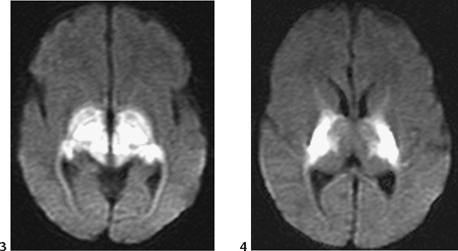
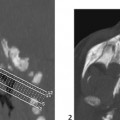
CASE 6 A neonate presents with an acute severe encephalopathy and odor of maple syrup (or burnt sugar). Figure 6A Figure 6B CT of the posterior fossa displays low attenuation of the cerebellar white matter, brachium pontis, and tegmentum pontis, as well as of the corticospinal fascicles of the anterior pons, with effacement of the fourth ventricle (Fig. 6A1). The midbrain and the cerebral hemispheres appear swollen also with diffusely low attenuation (Fig. 6A2). Diffusion-weighted MRI (DWI) of the posterior fossa shows a marked increase of signal of the cerebellar white matter, brachium pontis, tegmentum pontis, and corticospinal tracts (Fig. 6B1). The midbrain is swollen, with increased DWI signal of the superior cerebellar peduncles, of the central midbrain including the red nuclei, and of the lateral part of the pes pedunculi, as well as of the optic tracts (Fig. 6B2). The level above illustrates the involvement of the lateral geniculate bodies (Fig. 6B3), whereas the cut passing through the basal ganglia demonstrates involvement of the posterior limbs of the internal capsules bilaterally, as well as of the adjacent globi pallidi, and to some degree, of the anterior-lateral thalamic nuclei (Fig. 6B4). Maple syrup urine disease (MSUD; also called leucinosis) MSUD, also referred to as leucinosis, is a disorder that was first reported in 1954 in a family that lost four infants within the first 3 months of their lives because of a neurodegenerative disorder. The urine of these infants had an odor resembling that of maple syrup. MSUD is a branched-chain amino-acidopathy caused by a decreased activity of branched-chain a-ketoacid (BCKA) dehydrogenase, the second enzyme in the pathway for the degradation of leucine, isoleucine, and valine. The disease is mostly due to the increased concentration of leucine, as isoleucine and valine present little toxicity. This defect is rare in the general population (1/850,000) but may be common in some closed communities such as Mennonite settlements (up to 1/170). There are two forms of the disease. Both may be expressed in the same family. The classic form is characterized by an acute neonatal presentation, a low leucine tolerance, and a very low enzyme activity (<2.5% of controls). The variant phenotypes are less severe with a late onset, a reasonable leucine tolerance, and a fair enzyme activity (from 7 to 20% of controls), but variant forms also may develop acute deterioration. MSUD is an autosomal recessive disorder. There are now more than 50 different mutations known in genes, which govern the enzyme components of branched-chain amino acids (BCAAs) and BCKA dehydrogenase complex.
Clinical Presentation
Radiologic Findings
Diagnosis
Differential Diagnosis
Discussion
Background
Etiology
Pathophysiology
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


