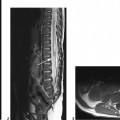
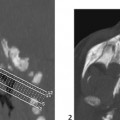
CASE 7 A 12-year-old child presents with bilateral deafness. Figure 7A Figure 7B The contrast-enhanced T1-weighted images of the head with fat saturation exhibit numerous lesions. On the axial plane through the internal auditory canals (IACs; Fig. 7A1), three schwannomas were identified. The largest one has developed from the right vestibular nerve; it occupies the whole IAC (flared) and the cerebellopontine angle (CPA) cistern, and encroaches on the middle cerebellar peduncle. On the left, another schwannoma is noted within the IAC. Two such lesions are sufficient for the diagnosis of neurofibromatosis type 2 (NF2). A smaller, third lesion is seen on the cisternal segment of the left abducens nerve. On the axial plane, through the temporal fossaeand orbits (Fig. 7A2), another lesion is seen curving around the right clinoid, with a large insertion on the dura typical for a meningioma. On the coronal plane passing through the CPA cisterns (Fig. 7A3), the two vestibular schwannomas are seen again in the IACs, as well as a broad-based meningioma inserted on the dura of the right foramen lacerum. Another, thinner meningioma is seen on the falx cerebri. Imaging of the spine in patients with NF2 is mandatory. On the coronal T1-weighted, fat-saturated image of the lumbar spine (Fig. 7B), schwannomas are seen at nearly every level in the intervertebral foramina, extending toward the periphery. Neurofibromatosis type 2 (NF2) Neurofibromatosis type 1 (NF1) for decades has been confused with NF2, although the expression of the two diseases is quite different (Table 7-1) with clearly identified different genetic defects. Both are tumor-producing diseases. NF1 is the most common genetic disease. It is characterized clinically by multiple, large, conspicuous “café au lait” spots, and by the presence of ocular Lisch nodules. Tumors in NF1 appear early in life. The most typical CNS tumors are the juvenile pilocytic astrocytoma (JPA) and the peripheral nerve sheath (PNS) neurofibroma. JPAs develop mostly on the anterior optic pathways, the tuber cinereum, and the basal ganglia/thalami. They are less aggressive than the sporadic ones, may enhance or not, or only partially, and tend to become dormant over the years. Neurofibromas may be proximal, paraspinal, peripheral, or cutaneous. As opposed to schwannomas, neurofibromas develop not from the sensory root but from the nerve. A strikingly complex form of neurofibroma is the plexiform neurofibroma that extends inextricably along the network of nerves into the soft tissues. On MRI and CT, neurofibromas enhance following contrast administration, but large masses may remain nonenhanced centrally. Rarely, neurofibromas may degenerate into neurofibrosarcomas, especially after radiation therapy. Other hallmarks of the disease are the bright signal spots (so-called hamartomas) that may be observed asymmetrically in the basal ganglia, posterior thalami, brain stem, and central cerebellum. These usually disappear at adulthood. Other brain tumors may rarely occur, as well as aqueductal stenosis. The last feature of NF1 that is never observed in NF2 is the mesenchymal dysplasia that affects the base of the skull (dysplasia or agenesis of the greater wing of the sphenoid, often associated with an adjacent plexiform neurofibroma), the spine (vertebral scalloping), the long bones, the dura (multiple lateral meningoceles), and the arteries (dysplastic giant aneurysms, moyamoya disease).
Clinical Presentation
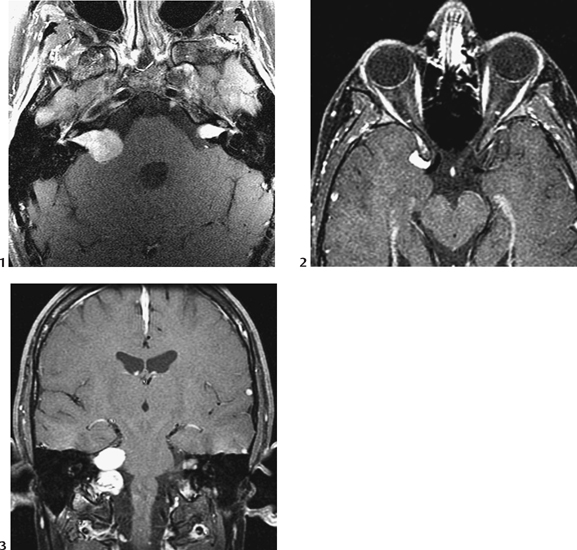
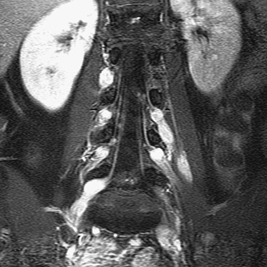
Radiologic Findings
Diagnosis
Differential Diagnosis
| Features | NF1 | NF2 |
| Incidence | 1/3000 | 1/50,000 |
| Chromosomal anomaly | Long arm 17 | 22q11 |
| Age at first symptoms | Early years | 2nd decade |
| Optic/hypothalamic JPA | Common | None |
| “Unidentified” bright spots | Common | None |
| Neurofibromas, paraspinal | Common | None |
| Neurofibromas, peripheral | Common |