Axial T2WI through the basal ganglia.
Multiple System Atrophy-Parkinsonian Type
Primary Diagnosis
Multiple system atrophy-Parkinsonian type
Differential Diagnoses
Parkinson disease
Multiple system atrophy-cerebellar type
Parkinson-plus syndrome
Corticobasal degeneration
Imaging Findings
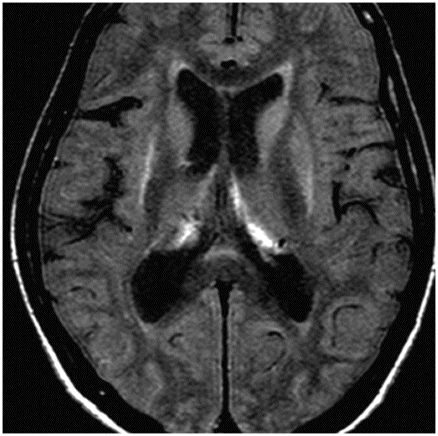
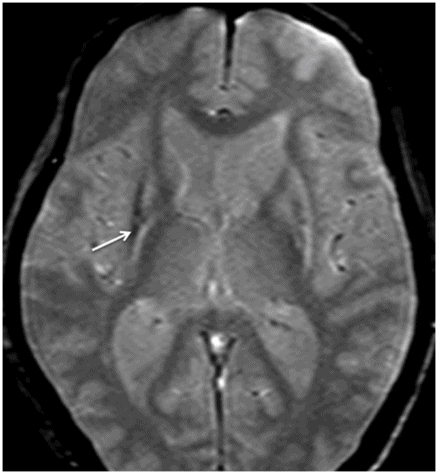
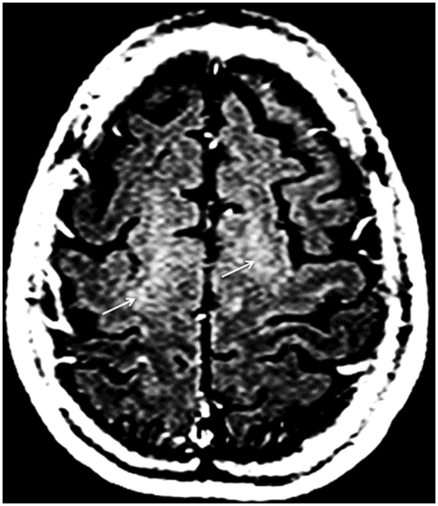
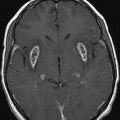
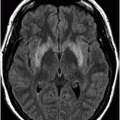
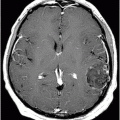
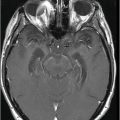
Fig. 9.1: Axial T2WI through the basal ganglia demonstrated subtle curvilinear T2 hyperintensity along the dorsolateral margins of the putamen (arrows) – the hyperintense putaminal slit sign. Putaminal hypointensity, which sometimes can be seen because of abnormal iron accumulation, is not present in this particular case. Fig. 9.2: Axial FLAIR image through the basal ganglia better demonstrated the hyperintense putaminal slit sign. Fig. 9.3: Axial GRE image through the basal ganglia demonstrated subtle hypointensity in the posterior aspect of the right putamen, due to abnormal iron deposition (arrow) that was not evident on the T2WI sequences. Fig. 9.4: Axial spin echo T1WI sequence with added magnetization transfer pulse demonstrated hyperintensity involving the corticospinal tract at the level of the centrum semiovale (arrows).
Discussion
Typically, Parkinson disease responds well to levodopa. Levodopa-unresponsive Parkinsonism suggests one of the atypical or Parkinson-plus syndromes, namely multiple system atrophy (MSA), corticobasal degeneration (CBD), or progressive supranuclear palsy (PSP). The presence of autonomic failure (urinary problems and erectile dysfunction) is suggestive of MSA. The absence of prominent ataxic signs, cerebellar and pontine atrophy, and hot-cross-bun sign disfavors diagnosis of MSA-C. This patient did not have eye movement problems, and his MRI did not show foreshortening of the midbrain, thus PSP can be eliminated as a potential diagnosis. The absence of significant brain atrophy, predominant behavioral symptoms, or cognitive decline eliminates CBD as an optional diagnosis.
Multiple system atrophy is a gradually worsening, neurodegenerative disorder characterized by a combination of levodopa-nonresponsive abnormal movements and symptomatic autonomic nervous system failure. As movement disorder is the predominant presenting symptom in all patients with MSA, it is considered one of the Parkinson-plus syndromes, in addition to PSP and CBD. Depending upon the predominant brain regions involved and subsequent clinical symptoms, MSA can be categorized as Parkinsonian type (MSA-P) or cerebellar type (MSA-C, see Part I: Case 8). There is an ethnic difference in the incidence of MSA-C verses MSA-P. MSA-P is more prevalent in North American and European populations, as compared to MSA-C, which is more prevalent in the Japanese population.
Multiple system atrophy is now classified as one of the alpha-synucleinopathies, due to the presence of cytoplasmic inclusion bodies that contain alpha-synuclein. However, no specific mutation of the synuclein (SNCA) gene has been implicated as a cause of MSA although several genetic polymorphisms at the SNCA locus have been linked to MSA. Recently, homozygous or compound heterozygous mutations in the COQ2 gene (which encode enzymes involved in the biosynthetic pathway of coenzyme Q10) have been linked to familial MSA.
Common presentation of MSA-P includes decreased spontaneous movement, tremor, or rigid muscles, clumsiness, loss of balance, and frequent falls. In addition, patients also demonstrate features of autonomic nervous system dysfunction manifested as fainting or lightheadedness due to orthostatic hypotension, and bladder control problems, such as a sudden urge to urinate or difficulty emptying the bladder completely. Although patients may initially demonstrate MSA-P symptoms, over time, they may develop MSA-C symptoms. Moreover, common presentations of MSA also include development of contractures in the hands and feet, disproportionate antecollis, emotional lability such as inappropriate laughing or crying, as well as sleep and sexual disturbances.
Unlike MSA-C, there is no characteristic imaging appearance associated with MSA-P. On conventional imaging, there may be posterior putaminal hypointensity from abnormal iron accumulation, which can be better seen on 3T magnets, or subtle hyperintensity at the dorsolateral margin of the putamen on T2-weighted sequences, which has been described as hyperintense putaminal rim or slit sign. Volumetric segmentation of putaminal volume has been shown to help differentiate MSA-P from controls and Parkinson disease. Although these are helpful indicators, these changes are not specific to MSA-P. Advanced MRI techniques may add complementary information to the conventional MRI. Diffusion MRI imaging has shown low anisotropy of the motor cortex and increased diffusivity of putamen in MSA-P. T1 spin echo MRI with addition of magnetization transfer contrast pulse has shown that there is an increase in signal in the motor cortex as well as in the pyramidal tracts at the centrum semiovale, particularly if MSA-P is associated with C9orf32 mutation associated amyotrophic lateral sclerosis. Striatal and cerebellar hypometabolism on 18F-fluorodeoxyglucose PET scan in a patient with clinically absent ataxia is suggestive of MSA-P.

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


