Adrenal lesions
Percent of total (n = 2005)
Functioning adenoma
11.4
Subclinical Cushing’s syndrome
5.3
Aldosterone-producing adenoma
1.0
Pheochromocytoma
5.1
Malignant
7.2
Adrenal cancer
4.7
Metastasis
2.5
Total potentially surgical lesions
18.6
Apparent nonfunctioning adenoma
82.4
Size and Growth
Median size of an adrenal incidentaloma at presentation is approximately 3 cm [15, 20]. Malignant potential increases with size [4, 15, 21], and adrenocortical carcinoma (ACC) accounts for 2 % of tumors up to 4 cm in size, 6 % of tumors that are 4.1–6 cm, and 25 % of tumors that are greater than 6 cm [16, 22]. In contrast, benign adrenal adenomas account for less than 15 % of masses greater than 6 cm [16]. Benign to malignant ratio has been estimated to be 5–1 5:1 at a cutoff of greater than 3 cm in diameter and 3–1 3:1 at a cutoff of greater than 4 cm [15]. While masses greater than 6 cm should be considered malignant until proven benign and generally require surgical excision [2, 16], many experts recommend 4 cm as the cutoff diameter for surgical resection in otherwise healthy patients [2, 13, 20, 23]. The 4 cm criterion has a 93 % sensitivity and 42 % specificity for detecting malignancy and is felt to minimize number of unnecessary operations while avoid missing virtually all ACC [20, 23]. However, ACC is more commonly found in younger patients and the incidence of benign adrenal adenomas increases with age [13, 20]. Therefore, the decision regarding adrenalectomy for an incidentaloma should integrate the assessment of metabolic activity, tumor size, imaging characteristics, patient age, comorbidity status, surgical risk, presence of other malignancies, and tumor growth kinetics [2, 4, 13].
A minority of adrenal masses grow over time, with approximately 9 % growing more than 1 cm in a mean period of 3 years [15]. Even in the group of growing masses, the rate of malignant transformation is exceedingly low at around 1 in 1000, with rapid growth as a worrisome sign [13, 15, 24]. Growth by 1 cm or more during the period of observation has been recommended as a criterion to consider surgical removal [4, 13, 15]. The current recommendations for for repeat imaging are at 6, 12, and 24 months [2, 4, 13, 16, 25]. Suspicious and unusual masses may require more frequent imaging protocol, while small (<2 cm), uniform, hypodense nodules in patients with no history of malignant disease may be monitored less frequently [13].
Biopsy of Adrenal Masses
Adrenal biopsy has limited role in the workup of adrenal masses. While biopsies have high positive and negative predictive values in patients with a known non-adrenal malignancy [26, 27], they cannot reliably differentiate benign from malignant adrenocortical tumors [2, 4, 13, 16, 23]. Although complication rate for adrenal biopsies is low, some complications, such as needle track seeding, hemorrhage, pneumothorax, and hemothorax, can seriously affect patient well-being and limit surgical options [2, 4, 13, 23]. Therefore, adrenal biopsy should be pursued only when its result is certain to influence management and radiologic and metabolic diagnostic limitations have been reached [2, 4, 13]. Prior to performing an adrenal biopsy, the possibility of pheochromocytoma must be evaluated with metabolic testing in order to prevent life-threatening hypertensive crises [16, 25].
Adrenal Mass Imaging
Computed tomography (CT) and magnetic resonance imaging (MRI) are able to discriminate with high degree of accuracy among the various types of incidentally detected adrenal lesions [3], but ultrasonography is suboptimal for detection and characterization of adrenal tumors [28, 29]. Important attributes, such as size, laterality, homogeneity, density, and anatomic relationships, are also accurately assessed by MRI and CT [2]. Large size, irregular shape, vague contour, and invasion into surrounding structures generally indicate malignancy [29]. However, metastatic lesions can be homogeneous and similar in appearance to adenomas [30]. Characterization of adrenal lesions relies on the identification of intracellular lipid, perfusion differences, and metabolic activity that differentiate a benign adenoma from nonadenomas [30, 31]. Since most (>70 %) adrenal adenomas contain much higher intracellular lipid content than other adrenal lesions, lipid sensitive imaging is highly effective in their identification [30, 32]. Nevertheless, CT or MRI characteristics do not eliminate the need for a complete metabolic workup of all adrenal tumors [2, 16].
Computed Tomography
The ideal adrenal CT should include unenhanced, 60 s postcontrast, and 15 min postcontrast images [2, 3], but an unenhanced CT is perhaps the single best and most easily interpretable test to diagnose an adrenal adenoma [2]. Adrenal adenomas demonstrate a low attenuation value on unenhanced CT due to their high lipid content. The measurement of attenuation value should involve a region of interest (ROI) that is one-half to two-thirds of the lesion’s surface area through the center of the lesion and avoid areas of necrosis [3, 30]. A Hounsfield unit (HU) of 0 or less is 100 % specific but only 47 % sensitive for a benign adenoma [33]; an attenuation value of 10 HU or less, which is currently the most widely adopted threshold, improves the sensitivity to a clinically acceptable 71 % while maintaining specificity of 98 % [30, 34]. In order to improve the sensitivity for detection of adenomas on unenhanced CT, some have advocated use of histogram analysis, where individual attenuation values of all pixels in the ROI are plotted against their frequency, and the amount of lipid in the mass is proportional to the number of negative pixels (less than 0 HU) [3, 30]. Up to 91 % sensitivity with threshold of >10 % negative pixel has been demonstrated [35, 36], but its utility in clinical practice remains unproven [30]. Unlike an unenhanced CT, the utility of a single contrast-enhanced (CE) CT is limited as tremendous overlap in postcontrast attenuation values exists between adenomas and nonadenomas [37].
Up to 30 % of adrenal adenomas are lipid-poor and have an attenuation of greater than 10 HU on unenhanced CT, making them indistinguishable from other lesions [34]. They demonstrate rapid washout of contrast, a property that is unique to adenomas [3, 29, 30]. While the absolute attenuation measurement on a delayed CT is not useful, the ratio of adrenal attenuation comparing washout-delayed scan to the initial dynamic enhanced scan is able to characterize adrenal lesions with great precision [30]. If an unenhanced CT has been obtained, the percentage of absolute contrast enhancement washout (ACEW) can be calculated by [3, 30]:
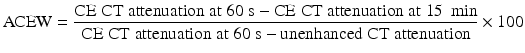
Frequently in clinical practice, the unenhanced CT is unavailable. The relative contrast enhancement washout (RCEW) is calculated in these scenarios [3, 30]:
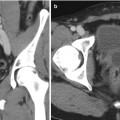
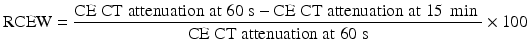 An ACEW of >60 % and a RCEW of >40 % after 15 min delay are both highly sensitive and specific for an adenoma (sensitivity 88 and 96 %, specificity 96 and 100 %, respectively), and these thresholds are adopted most commonly [3, 30, 38] (Fig. 26.1). Although a >50 % RCEW after 10 min has been reported to have a high sensitivity and specificity [30, 39], the 10 min protocol was later found to lack sufficient accuracy for routine clinical practice [40].
An ACEW of >60 % and a RCEW of >40 % after 15 min delay are both highly sensitive and specific for an adenoma (sensitivity 88 and 96 %, specificity 96 and 100 %, respectively), and these thresholds are adopted most commonly [3, 30, 38] (Fig. 26.1). Although a >50 % RCEW after 10 min has been reported to have a high sensitivity and specificity [30, 39], the 10 min protocol was later found to lack sufficient accuracy for routine clinical practice [40].
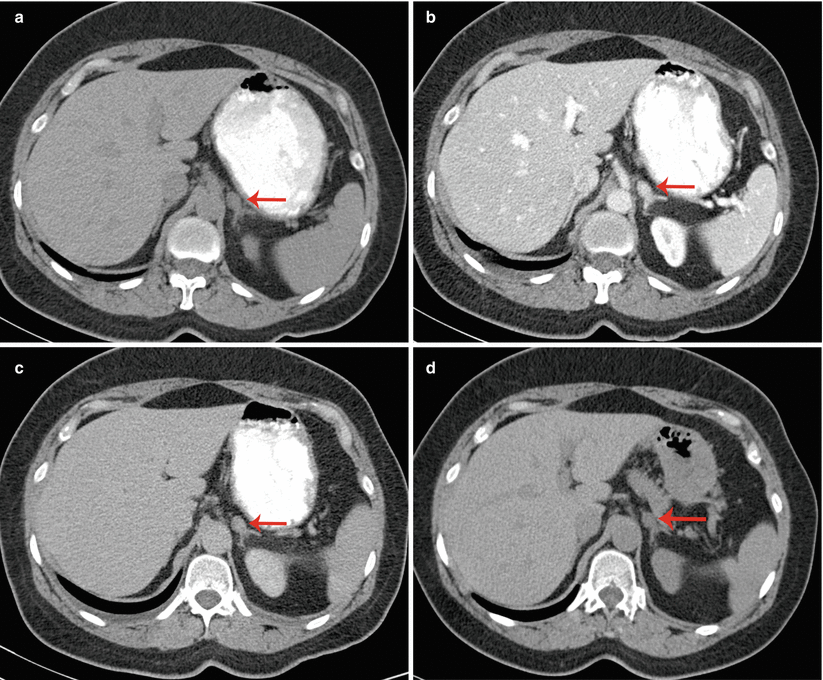
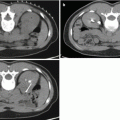
Fig. 26.1
A 53-year-old woman underwent an adrenal protocol CT for evaluation of a left adrenal nodule found during workup of hyperaldosteronism. (a) Unenhanced CT shows a left adrenal nodule (arrow) with an attenuation value of 14 HU, indicating an indeterminate mass. (b) Delayed images 60 s after contrast injection show that the mass has an attenuation value of 142 HU (arrow). (c) Delayed images 15 min after contrast injection show that the mass has an attenuation value of 60 HU (arrow). The mass is consistent with an adenoma with an absolute contrast enhancement washout of 64 % and relative contrast enhancement washout of 58 %. (d) Unenhanced CT 6 months later shows unchanged mass size (arrow)
Magnetic Resonance Imaging
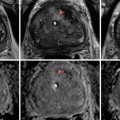
Standard adrenal MRI should include T1, T2, chemical shift imaging (CSI) in- and opposed-phase, and T1 fat-saturated sequences [3]. The normal adrenal has a uniform intermediate T1 and a low-to-intermediate T2 signal intensity, and it is better demonstrated on T1 fat-saturated images due to the suppression of signal from surrounding retroperitoneal fat [3]. CSI detects intracellular lipid in adenomas by exploiting the different frequency of proton oscillation in water and lipid molecules [3, 30, 41]. In adenomas that contain intracellular lipid, the signal intensity on opposed-phase sequences drops out compared to in-phase images, whereas it remains unchanged in malignant lesions and pheochromocytomas, which lack intracellular lipid [3, 30, 32, 41] (Fig. 26.2). In general, simple visual assessment of relative signal intensity loss is sufficient, but equivocal cases may need quantitative evaluation [3, 41, 42]. The adrenal-to-spleen chemical shift ratio of <70 or signal intensity index of >16.5 % may be used to identify lipid-rich adenoma [3, 30, 42, 43]. In lipid-rich adenoma, CSI is similar to unenhanced CT in accuracy and avoids ionizing radiation, although at significantly higher cost [30]. However, in lipid-poor adenomas, particularly those with unenhanced CT attenuation values between 10 and 30 HU, CSI appears to be superior [44, 45]. Further modification of CSI to subtraction CSI, where the opposed-phase images are subtracted from the in-phase images, can be helpful in equivocal cases to further improve diagnostic accuracy [46]. Unlike contrast-enhanced CT washout studies, gadolinium-enhanced MR washout studies do not exhibit sufficient diagnostic strength [2–4]. Nevertheless, adenomas demonstrate certain enhancement patterns that may aid in discriminating them from malignant lesions, such as homogeneous enhancement, ring enhancement, and early peak enhancement [47].
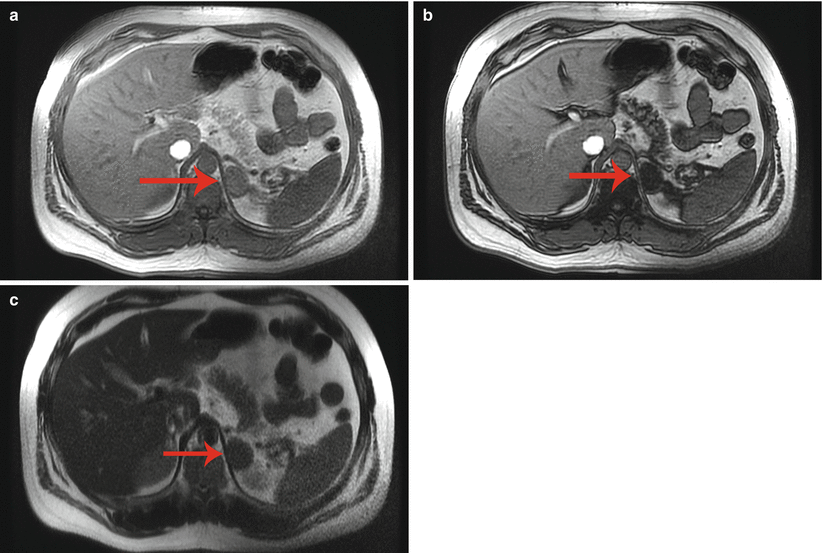
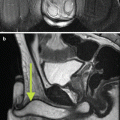
Fig. 26.2
A 45-year-old man underwent a 1.5 T MRI with CSI that was consistent with left adrenal adenoma. (a) In-phase T1-weighted imaging demonstrates a left adrenal mass (arrow) with signal isointense to muscle. (b) Opposed-phase T1-weighted imaging shows dropout of signal in the left adrenal lesion (arrow) relative to the in-phase imaging. (c) Single-shot T2 weighted spin echo imaging reveals the left adrenal nodule (arrow) with low signal intensity
Functional Imaging
18F-fluorodeoxyglucose positron emission tomography (18F-FDG-PET) has demonstrated high sensitivity and specificity in identification of malignant adrenal tumors, including adrenocortical carcinomas, metastatic pheochromocytomas, and metastatic lesions to the adrenal gland [48–50]. 11C-metomidate, a radiotracer that is preferentially taken up by adrenocortical cells, is used to distinguish adrenocortical and non-cortical lesions, but its use is significantly limited by its inability to differentiate benign adenomas from adrenocortical carcinomas [51–53].
Radionuclide scintigraphy with 131I-6β-iodomethyl-norcholesterol (NP-59), another marker of adrenocortical tissue, can distinguish lesions of adrenocortical origin from other space-occupying lesions [17]. However, NP-59 is also unable to identify adrenal adenomas from adrenocortical carcinomas, and it is currently not approved by the US Food and Drug Administration [2, 3, 30]. 131I or 123I metaiodobenzylguanidine (MIBG) is an analogue of norepinephrine that is incorporated into catecholamine storage vesicles and is used to identify pheochromocytomas [29, 54]. However, MIBG scintigraphy has suboptimal sensitivity and specificity, particularly in extra-adrenal, malignant, and certain familial diseases, limiting its utility [2, 50]. Overall, the indication for functional imaging in the workup of adrenal incidentalomas is restricted due to the high diagnostic accuracy of biochemical evaluation and cross-sectional imaging [2, 4, 25, 30].
Biochemical Evaluation
More than 10 % of adrenal incidentalomas are metabolically functional [19]. Metabolic testing for cortisol and catecholamine is recommended for all adrenal incidentalomas [16]. Aldosterone hypersecretion should also be assessed in patients with a history of hypertension [13, 16, 55]. Since sex steroid hypersecretion is exceedingly rare, routine testing for sex hormones is not recommended [13, 16].
Testing for Cortisol Hypersecretion
The three initial tests for Cushing’s syndrome in patients with adrenal incidentalomas are overnight low-dose (1 mg) dexamethasone suppression test (1-mg DST), late-night salivary cortisol test, and 24-h urinary free cortisol test (UFC). Practice guidelines from the Endocrine Society recommend the 1-mg DST or late-night salivary cortisol test over UFC due to their superior sensitivity in this population [56].
For the 1-mg DST, 1 mg dexamethasone is given between 2300 and 2400 h on the night before, and serum cortisol is obtained between 0800 and 0900 h. Alternatively, some endocrinologists prefer a 48 h, 2-mg/day DST for potential improved sensitivity. Patient’s failure to suppress cortisol level is indicative of Cushing’s syndrome [56]. A cutoff value of >1.8 μg/dL, with a sensitivity of >95 % and specificity of 70–80 %, is recommended for this screening test [56]. Various pharmaceuticals may affect DST via their effect on CYP 3A4, such as phenytoin, phenobarbital, carbamazepine, rifampin, and alcohol. Therefore, patients receiving antiepileptic medications known to affect dexamethasone metabolism should not undergo DST [56]. Oral contraceptives should be avoided since its estrogen component causes high false-positive rates by increasing cortisol-binding globulin concentration and thus total (but not bioavailable) cortisol levels [56].
Late-night salivary cortisol examines the loss of circadian rhythm in Cushing’s syndrome [56]. Measurements collected between 2300 and 2400 h with normal cutoff of <145 ng/dL have sensitivity and specificity of >90 % [56]. This test may be influenced by patient factors including depression, altered sleep patterns, chronic illness, and tobacco use [56].
Twenty-four-hour urinary free cortisol evaluation directly measures free bioavailable cortisol. This test is most accurate in patients with a glomerular filtration rate >60 mL/min. It is the recommended test in pregnant patients, although cutoff values two to three times the normal threshold must be used to account for the physiologic hormonal changes [56].
Testing for Aldosterone Hypersecretion
Although hypersecretion of aldosterone by adrenal masses is rare, with only 1 % adrenal incidentalomas causing Conn syndrome [19], nearly 5 % newly hypertensive patients referred to hypertension centers have been found with aldosterone-producing adenoma [57]. The screening test of choice for aldosterone hypersecretion is a morning plasma aldosterone (ng/dL) to renin (ng/mL/h) ratio, where a ratio >20 or 30 and simultaneous aldosterone concentration >15 ng/dL are indicative of Conn syndrome [13, 16, 58, 59]. Patients with hypokalemia should normalize their potassium levels [13]; patients on beta blockers, potassium-sparing diuretics such as amiloride, and mineralocorticoid receptor blockers such as spironolactone should stop those medications to prevent false results [13, 59]. Confirmatory testing is necessary for patients with positive screening test, usually with a 3-day oral sodium challenge followed by 24-h urinary aldosterone measurement, and endocrinology referral should be considered [2, 13, 59, 60].
Testing for Catecholamine Hypersecretion
Approximately 5 % of adrenal incidentalomas are pheochromocytoma [19]. Biochemical testing to rule out this disease should be performed in normotensive as well as hypertensive patients, as about half of patients with pheochromocytoma are not hypertensive [20, 61]. Either plasma-free metanephrine or 24-h urinary-fractionated metanephrine tests should be the initial biochemical testing for pheochromocytoma [50, 62, 63]. The superiority of the two tests has not been established [50], and both provide sensitivity of 97 % and specificity around 90 % [64, 65]. Inherent characteristics of each test should be considered when choosing between the two. For example, plasma-free metanephrine is applicable in patients with renal failure, where urine collection may be inappropriate [50, 62, 63]. Conversely, it is recommended that blood be sampled after patient has been supine for 30 min, thus requiring more time and effort from the medical staff [50, 62, 63]. A number of medications, including acetaminophen, tricyclic antidepressants, phenoxybenzamine, monoamine oxidase inhibitors, and α-methyldopa, cause false-positives and should be avoided [50]. Physiologic stress associated with extreme illness can also elevate plasma or urine metanephrines [50]. If results are equivocal, repeat testing after elimination of confounding medications, sampling errors, and physiologic stress should be performed. If abnormal results continue, referral to an endocrinologist is recommended [2, 50], and additional investigation, such as clonidine suppression test and chromogranin measurements, may be performed [50].
Adrenal Diseases
Cushing’s Syndrome
Cushing’s syndrome is defined as inappropriately high exposure of tissue to glucocorticoids [56]. Hypercortisolism can cause diverse effects on different organ systems (Table 26.2), and some physical features include reddish purple striae, plethora, proximal muscle weakness, and easy bruising [56]. Causes of Cushing’s syndrome can be divided into exogenous, due to iatrogenic glucocorticoid administration, and endogenous, which is further separated into ACTH-dependent and ACTH-independent [2, 56]. The most common cause of Cushing’s syndrome is use of supraphysiologic amounts of exogenous glucocorticoids [66]. Of the endogenous Cushing’s syndrome, ACTH-dependent hypercortisolism accounts for 80–85 %, with Cushing’s disease, or primary pituitary hypersecretion of ACTH, being the most common cause [66]. Ectopic ACTH production nearly always arise from malignant processes, most commonly from small-cell carcinoma of the lung and bronchial carcinoid tumors [10, 66, 67]. An extremely rare cause is production of corticotrophin-releasing hormone (CRH) by malignancies, most commonly bronchial carcinomas [67]. ACTH-independent Cushing’s syndrome is most often caused by unilateral adrenal tumors, including benign adenomas and adrenocortical carcinomas, in approximately 60 and 40 % of cases, respectively [66]. Other rare causes of Cushing’s syndrome include ACTH-independent bilateral macronodular adrenal hyperplasia, as seen in McCune-Albright syndrome, and primary pigmented nodular adrenocortical disease, as seen in Carney complex [66–68].
Table 26.2
Effects of glucocorticoids
System | Effect |
|---|---|
Central nervous system | Depression Psychosis |
Endocrine system | Decreased LH, FSH release Decreased TSH release Decreased GH secretion Menstrual irregularity |
Eyes | Glaucoma |
Cardiovascular/renal systems | Salt and water retention Hypertension |
Gastrointestinal tract | Peptic ulcerations |
Carbohydrate/lipid metabolism | Increased hepatic glycogen deposition Increased peripheral insulin resistance Increased gluconeogenesis Increased free fatty acid production Type 2 diabetes |
Adipose tissue distribution | Visceral obesity |
Genitourinary system | Urolithiasis Erectile dysfunction Decreased libido |
Bone and calcium metabolism | Vertebral osteoporosis |
Skin/muscle/connective tissue | Protein catabolism/collagen breakdown Skin thinning Muscular atrophy Buffalo hump Hirsutism or female balding Acne |
Immune system | Anti-inflammatory actions Immunosuppression |
Growth and development | Decreased linear growth |
After diagnosing Cushing’s syndrome (see section “Testing for Cortisol Hypersecretion”), the cause of hypercortisolism should be determined with endocrinologic referral [56, 66]. ACTH-independent causes are distinguished from ACTH-dependent ones by a low serum ACTH, and abdominal imaging is employed to identify the adrenal source [66]. For ACTH-dependent Cushing’s syndrome, direct downstream measurement of ACTH in the inferior petrosal sinus following CRH stimulation is the gold standard for separating ectopic ACTH production from Cushing’s disease [66, 67].
The main treatment option for Cushing’s disease is transsphenoidal surgical resection, and salvage radiation therapy is reserved for persistent or recurrent cases [66, 67]. For unilateral cortisol-producing adrenal masses, laparoscopic resection is preferred [66, 67]. Bilateral adrenalectomy with lifelong replacement therapy may be pursued for bilateral hyperplasia or ACTH-dependent causes that are not amenable to surgical resection of the source of ACTH [66]. For patients who are not candidates for surgical intervention, medical blockade with metyrapone, ketoconazole, mitotane, or etomidate may be attempted, although it is associated with significant toxicity [10, 66, 69, 70].
Primary Hyperaldosteronism
Primary hyperaldosteronism, or Conn syndrome, occurs in 5–13 % of hypertensive patients [22, 60]. In this disease, aldosterone secretion is independent of the renin-angiotensin-aldosterone system and suppresses plasma renin levels [2, 71]. Patients are usually diagnosed in the third to sixth decades of life [2, 71]. Signs and symptoms include refractory hypertension, hypokalemia, periodic paralysis, muscle weakness and cramping, polydipsia, and polyuria [60, 71]. However, no characteristic is specific to this syndrome, and hypokalemia is frequently absent [60, 71]. The vast majority of primary hyperaldosteronism is caused by bilateral adrenal hyperplasia (60 %) and adrenal adenoma (35 %) [60]. Comparatively, aldosterone-producing adenomas are associated with more profound hypertension and hypokalemia [57]. Less common causes of primary hyperaldosteronism include unilateral adrenal hyperplasia (2 %), adrenocortical carcinoma (<1 %), familial hyperaldosteronism (<1 %), and ectopic aldosterone-producing adenoma or carcinoma (<1 %) [60]. Conn syndrome is associated with earlier and worse cardiac and renal damage, underlying the need for treatment [72, 73].
Indications for primary hyperaldosteronism screening are listed in Table 26.3. Once screening and confirmatory testing achieve the diagnosis of the disease (see section “Testing for Aldosterone Hypersecretion”), subtype differentiation is essential to determine surgically correctable disease, including aldosterone-producing adenoma or carcinoma, primary unilateral adrenal hyperplasia, or ectopic aldosterone-producing tumors [2, 55]. An adrenal CT scan is indicated to evaluate for presence of adrenal nodules in potential surgical candidates [2]. Correct lateralization is more important with advancing age as the incidence of adrenal incidentalomas increases [55]. A young patient (<40 years old) with primary hyperaldosteronism who has an adrenal adenoma >1 cm and a morphologically normal contralateral adrenal gland may proceed with surgical resection of the adenoma without need for further imaging given the rarity of incidental adenomas in this population [2, 55, 60, 74, 75]. Patients considering surgery who are older, with bilateral abnormal glands or with unilateral microadenoma(s), should undergo bilateral adrenal vein sampling for lateralization of aldosterone secretion [2, 55, 74, 76]. The details of adrenal vein sampling will be discussed in the next chapter.
Table 26.3
Indications for primary hyperaldosteronism screening
Hypertension with hypokalemia |
Resistant hypertension |
Adrenal incidentaloma with hypertension |
Early onset hypertension (<20 years) or stroke (<50 years) |
Severe hypertension (≥160 mmHg systolic or ≥110 mmHg diastolic) |
Whenever considering secondary causes of hypertension |
Unexplained hypokalemia (spontaneous or diuretic induced) |
Evidence of target organ damage disproportionate to degree of hypertension |
Treatment for primary hyperaldosteronism aims to remove the source of excess aldosterone or to block its effect on target organs [60]. Laparoscopic unilateral adrenalectomy is preferred for aldosterone-producing adenomas or unilateral adrenal hyperplasia [60, 74]. Although blood pressure control improves in 100 % of patients postoperatively, long-term cure rate of hypertension ranges from 30 to 72 %, most likely due to concurrent primary hypertension [60, 77, 78]. Medical therapy with aldosterone antagonists spironolactone or eplerenone is indicated for patients who have surgically uncorrectable subtypes and those who are not surgical candidates [60, 74].
Pheochromocytoma
Pheochromocytoma is a tumor arising from catecholamine producing adrenomedullary chromaffin cells [50]. Its prevalence in hypertensive patients in general outpatient clinics varies between 0.2 and 0.6 % [50]. Approximately 3–5 % of adrenal incidentalomas are pheochromocytomas [19, 20, 79]. About 10 % of pheochromocytomas are located bilaterally [59]. Up to a quarter of pheochromocytomas are extra-adrenal and are known as paragangliomas, as they arise from paraganglia that anatomically parallel the sympathetic and parasympathetic ganglia of the head, neck, thorax, abdomen, and pelvis [2, 50, 80]. Pheochromocytoma may be malignant, which can only be defined by the presence of clinical metastasis [2]. While less than 5 % of adrenal pheochromocytomas are malignant, metastases accompany a third of extra-adrenal diseases [81]. The 5-year survival rate of malignant pheochromocytoma is less than 50 % without treatment [82]. More than a third of patients have been found to harbor germline mutations [50]. The most common genes involved in germline mutations are mitochondrial succinate dehydrogenase subunit B and D (SDHB, SDHD), von Hippel-Lindau (VHL), rearranged transfection protooncogene (RET), and neurofibromatosis type 1 (NF1) [50] (Table 26.4). Of the familial forms of pheochromocytoma, mutations of SDHB lead to metastatic disease in more than 40 % of patients [50, 83–85].
Table 26.4
Hereditary forms of pheochromocytoma
Syndrome | Clinical characteristics | Gene | % of all PHEO patients | Risk of PHEO (%) | Rate of malignant disease (%) |
|---|---|---|---|---|---|
Familial paraganglioma syndrome type 4 | Carotid body tumors Vagal, jugular, tympanic, thoracic, or abdominal paragangliomas | SDHB | 10.3 | 20 | >40 |
Familial paraganglioma syndrome type 1 | Carotid body tumors Vagal, jugular, tympanic, thoracic, or abdominal paragangliomas | SDHD | 8.9 | 20 | <3 |
Von Hippel-Lindau syndrome | Renal cell carcinomas and cysts Brain and retinal hemangioblastoma Pancreatic neoplasms and cysts Epididymal cystadenomas Endolymphatic sac tumors | VHL | 7.3 | 10–20 | 5 |
Multiple endocrine neoplasia type 2A | Medullary thyroid cancer Primary hyperparathyroidism Cutaneous lichen Amyloidosis | RET | <5 | 50 | 3 |
Multiple endocrine neoplasia type 2B | Medullary thyroid cancer Mucocutaneous neuromas Skeletal deformities (Marfanoid body habitus) Joint laxity Myelinated corneal nerves Intestinal ganglioneuromas (Hirschsprung disease) | RET | <5 | 50 | 3 |
Neurofibromatosis type 1 | Neurofibromas Café au lait skin spots Malignant peripheral nerve sheath tumor
Related posts:Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|