In 2016, the World Health Organization (WHO) central nervous system (CNS) classification scheme incorporated molecular parameters in addition to traditional microscopic features for the first time. Molecular markers add a level of objectivity that was previously missing for tumor categories heavily dependent on microscopic observation for pathologic diagnosis. This article provides a brief discussion of the major 2016 updates to the WHO CNS classification scheme and reviews typical MR imaging findings of adult primary CNS neoplasms, including diffuse infiltrating gliomas, ependymal tumors, neuronal/glioneuronal tumors, pineal gland tumors, meningiomas, nerve sheath tumors, solitary fibrous tumors, and lymphoma.
Key points
- •
The 2016 update to the fourth edition of the World Health Organization central nervous system (CNS) tumor classification scheme represents a fundamental change in the manner in which tumors are classified and for the first time incorporates molecular parameters in addition to traditional microscopic features into the updated classification scheme.
- •
The most impactful changes involve classification of diffuse gliomas, with the incorporation of genetically defined features.
- •
Molecular markers of the genetically defined tumors are not only of diagnostic significance but also of prognostic value; examples include isocitrate dehydrogenase mutations and 1p/19q codeletions in astrocytomas and oligodendrogliomas, respectively.
- •
Imaging remains a mainstay modality in the diagnosis and management of adult primary CNS tumors, and familiarity with the new scheme is crucial for neuroradiologists to convey meaningful information to their referring colleagues.
Introduction
Updates in the 2016 World Health Organization Central Nervous System Tumor Classification
Primary central nervous system (CNS) tumors are the seventh most common adult neoplasm, with an overall incidence rate of 23 cases per 100,000 people in the United States. The most widely accepted classification scheme is based on the World Health Organization (WHO) classification of tumors of the CNS, currently in its fourth edition. For nearly the past century, the classification of CNS tumors was traditionally based on microscopic features, with the assumption that tumors could be classified according to histologic similarities based on the cells of origin (eg, astrocytomas originating from astrocytes or oligodendrogliomas originating from oligodendrocytes) and further subclassified based on their degree of cellular differentiation. The past 2 decades of genetic and epigenetic research, however, have clarified the basis of tumorigenesis in a manner that has fundamentally altered the classification system. In the 2016 WHO classification, an update to the fourth edition represents a major paradigm shift in the manner in which CNS tumors are classified, for the first time integrating molecular parameters (genotypic) into the traditional microscopic features (phenotypic).
The implications of this updated classification are both broad and deep. For example, prognoses of certain tumors often correlate more strongly with molecular markers than with histologic grade (eg, isocitrate dehydrogenase [IDH] mutation status). The presence of molecular markers adds a level of objectivity and reproducibility conspicuously missing in a diagnostic process solely dependent on microscopic observation (eg, oligodendroglioma). Reconciling discordance between genotypic and phenotypic parameters is now an issue. For example, a glioma that histologically and phenotypically appears astrocytic may possess a 1p/19q codeletion, a genotypic parameter that is almost always seen in oligodendrogliomas. Conversely, a tumor that histologically resembles oligodendroglioma may possess ATRX and TP53 mutations with an intact 1p and 19q, which are molecular signatures genotypically consistent with astrocytomas. Therefore, familiarity with the new classification scheme is critical for neuroradiologists to convey meaningful information for optimal collaborative disease management by radiation oncologists, neuropathologists, neuro-oncologists, and neurosurgeons.
Classification Considerations: Layered Diagnosis, Not Otherwise Specified, Not Elsewhere Classified
The concept of a layered diagnosis was introduced to assist in systematically diagnosing CNS tumors, incorporating both genotypic and phenotypic parameters into the diagnosis. Layer 1 represents the integrated diagnosis, whereby a unifying summation of the molecular and microscopic data most accurately represents the diagnostic entity and can be generated only if information from all other layers are present. Layer 2 represents the histologic classification (eg, oligodendroglioma). Layer 3 presents the WHO grade (eg, WHO grade III). Layer 4 represents the molecular parameters (eg, 1p/19q codeletion). A layer 2 and 3 diagnosis can be made when no molecular data are available and is used to convey that a full, integrated layer 1 diagnosis is not possible.
The suffix not otherwise specified (NOS) is a designation in the 2016 WHO classification that denotes that the necessary molecular assays were unavailable or not performed at the facility to provide a layer 4 diagnosis for entities that may qualify for an integrated layer 1 diagnosis (eg, oligodendrogliomas, astrocytomas, or embryonal tumors). Therefore, the suffix conveys through the pathology report that the case has not been worked up to the necessary extent for a full, integrated layer 1 diagnosis. In contrast, the suffix, not elsewhere classified (NEC), is a designation used when molecular data analyses are performed and available, but the histologic (layer 2 and 3) and molecular (layer 4) data altogether are conflicting, therefore preventing a consensus diagnosis. It also may be used for increasingly well-described genetically defined entities for which there is no official recognition by the WHO classification scheme. This suffix is not yet codified in the 2016 WHO classification but likely will be utilized in the upcoming fifth edition. For example, the NEC designation can be used in cases of histologically classic oligodendroglioma with an IDH-type molecular marker or histologically classic diffuse astrocytoma with molecular features of glioblastoma. NEC also can be used with new molecular entities, such as glioblastoma, FGFR3-TACC3 fusion, which is resistant to conventional chemoradiation. It becomes important for the neuropathologist to identify these gliomas for inclusion in targeted therapeutic trials.
Goals and Objectives
Although a comprehensive review of adult primary brain neoplasms based on the 2016 WHO classification is outside the scope of this article, the goal is to impart the readership with a working knowledge of the typical MR imaging findings of commonly encountered adult primary brain neoplasms and the relevant changes in 2016 WHO classification.
Imaging technique and considerations
Imaging plays a major role as it represents the modern-day gross specimen. A differential diagnosis in order of probability can be formulated by scrutinizing all sources of information from a patient’s clinical and demographic features (age, gender, and presenting symptoms/signs) to imaging findings: anatomic location, borders, and tissue characteristics of the lesion as well as the presence and pattern of contrast enhancement. A routine brain MR image offers conventional sequences used to evaluate brain tumors, including precontrast and postcontrast T1-weighted imaging (T1WI), T2-weighted imaging (T2WI), T2-weighted fluid-attenuated inversion recovery (FLAIR), diffusion-weighted imaging (DWI), and either gradient-recalled echo (GRE) or susceptibility-weighted imaging (SWI). Small field-of-view, thin-slice sagittal T2WI and sagittal postcontrast T1WI provide the best diagnostic value for tackling pineal region tumors. High-resolution T2WI, constructive interference in steady state, or fast imaging employing steady-state acquisition sequences of the cerebellopontine angle and internal auditory canal can be used to evaluate vestibular schwannomas. Small field-of-view, thin-slice precontrast and postcontrast images as well as postcontrast dynamic images are helpful when assessing sellar region neoplasms. Unless explicitly stated as otherwise, imaging findings discussed within this article are based on MR imaging.
Pathology and imaging
Diffusely Infiltrating Gliomas
Gliomas represent a diverse and heterogeneous group of tumors. Based on the traditional phenotypic classification scheme, gliomas were thought to arise from a glial cell of origin. Therefore, subtypes of glial cells, such as astrocytes and oligodendrocytes, give rise to specific types of glioma, astrocytoma and oligodendroglioma, respectively. Gliomas can be relatively circumscribed with a tendency toward benignity (eg, pleomorphic xanthoastrocytoma, pilocytic astrocytoma, and subependymal giant cell astrocytoma) or diffusely infiltrating with tendency toward malignancy (eg, diffuse astrocytoma and oligodendroglioma). Previously, in the 2007 WHO classification, all astrocytic tumors were grouped together. Due to differences in growth pattern and behavior between circumscribed and diffusely infiltrating gliomas, all diffusely infiltrating gliomas are now amassed into 1 category regardless of histologic subtype (eg, astrocytic or oligodendroglial). Based on the 2016 WHO classification, diffusely infiltrating gliomas are comprosed of diffusely infiltrating astrocytomas, glioblastomas, and oligodendrogliomas.
Diffuse and anaplastic astrocytoma
After histologic confirmation and grading of diffusely infiltrating astroctyomas, including WHO grade II diffuse astrocytomas (not to be confused with the general term, diffusely infiltrating astroctyomas) and WHO grade III anaplastic astrocytomas (grade III), these tumors are further stratified by IDH mutations. Approximately 90% of grades II and III diffusely infiltrating astrocytomas are IDH-mutants. Diffuse astrocytoma IDH–wild-type is such an uncommon diagnosis that reevaluation of an alternative diagnosis is often required. Anaplastic astrocytoma IDH–wild-type also is uncommon, with most tumors sharing genetic features with those of glioblastoma IDH–wild-type. Several recent studies have suggest minimal prognostic difference between grade II and grade III IDH-mutant astrocytomas and further suggest that IDH status serves as a more accurate prognostic marker (IDH-mutant more favorable than IDH–wild-type) than WHO grading. At this time, however, the WHO grading scheme along with IDH status is retained; amendments are likely to occur in the next revision.
On imaging, diffusely infiltrating astrocytomas are T2/FLAIR hyperintense masses that frequently arise from the cerebral hemispheres, with predilection for the frontal lobes. Despite their infiltrating growth pattern, they appear relatively circumscribed on imaging, tend to involve the underlying white matter, and are associated with characteristic cortical infiltration and gyral expansion. Both diffuse and anaplastic astrocytomas typically do not restrict on DWI and do not enhance with contrast. Enhancement is variably present in anaplastic astrocytomas ( Figs. 1 and 2 ). Ultimately, no distinctive imaging features reliably distinguish astrocytomas by WHO grade and IDH status.


Glioblastoma
Glioblastoma is a grade IV diffusely infiltrating astrocytoma that is also further stratified by IDH mutation status. Glioblastoma IDH–wild-type type arises de novo (clinically defined as primary glioblastoma), whereas glioblastoma IDH-mutant arises from a lower-grade IDH-mutant diffusely infiltrating astrocytoma (clinically defined as secondary glioblastoma). Unlike astrocytoma, approximately 90% to 95% of glioblastomas are IDH–wild-type, tend to occur in patients greater than 55 years old, and portend a worse prognosis compared with IDH-mutants. Despite arising de novo, glioblastoma IDH–wild-type shares other genetic alterations (eg, TERT promoter, EGFR amplification, and PTEN mutations) as well as clinical features with diffusely infiltrating astrocytoma IDH–wild-type, perhaps reflecting a spectrum of the same entity.
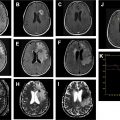
The term, multifocal , glioblastoma , often is used when referring to multiple, seemingly separate foci of tumor that are connected by spread of disease via white matter tracts or cerebrospinal fluid. On the other hand, the term, multicentric glioblastoma , often is used when there is no obvious imaging finding demonstrating a connection among separate tumor foci. Glioblastoma IDH–wild-type tumors classically demonstrate a thick, irregular rind of enhancing tumor, which histologically correlates with vascular proliferation. Centrally, a nonenhancing necrotic core typically preferentially involves the subcortical and deep white matter ( Fig. 3 ) on postcontrast T1WI. T2/FLAIR sequences demonstrate a poorly marginated, heterogeneously hyperintense mass with mass effect and extensive vasogenic edema. Tumor invariably extends beyond the regions of enhancement, even beyond the extent of visible peritumoral edema, frequently along white matter tracts; this is referred to as microscopic invasion. The moniker, butterfly glioma, has been used when symmetric corpus callosum involvement is present. Nonenhancing tumor and vasogenic edema are virtually indistinguishable radiologically. Tumor heterogeneity is often due to a combination of necrosis and intratumoral hemorrhage; the latter is common and easily elucidated on GRE or SWI. Calcification and restricted diffusion are rare.

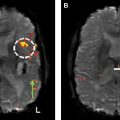
In contrast to astrocytomas, certain imaging findings may help distinguish glioblastoma IDH-mutants from IDH–wild-type. Glioblastoma IDH-mutants preferentially involve the frontal lobes and commonly have regions of nonenhancing tumor, not including the admixture of peritumoral edema/tumor. The thick enhancing rind, intratumoral hemorrhage, and large centrally necrotic components, which are classic features of glioblastoma IDH–wild-type, are infrequent features in glioblastoma IDH-mutants ( Fig. 4 ).

Oligodendroglioma
Oligodendroglioma (WHO grade II) and anaplastic oligodendroglioma (WHO grade III) share the same IDH gene family mutation as diffusely infiltrating astrocytomas. Oligodendrogliomas often are distinguished from astrocytomas by the presence of an additional 1p/19q codeletion. The less frequently encountered histologically classic oligodendroglioma with IDH–wild-type is given the diagnosis oligodendroglioma, NOS, after exclusion of other possible entities. In addition to its diagnostic value, the presence of a 1p/19q codeletion is prognostically significant in diffuse gliomas. Those with the codeletion receiving radiation followed by procarbazine, lomustine, and vincristine chemotherapy demonstrate significantly higher survival rates compared with those without the codeletion. In contrast, similar to astrocytomas, the prognostic value of grading remains an issue, because several studies have failed to identify a difference in survival by WHO grade after IDH mutation status stratification. Amendments to this histology-driven grading scheme may occur in the upcoming revision of the WHO classification scheme.
Oligoastrocytomas were previously diagnosed based on the histologic admixture of both astrocytic and oligodendrolglial components; however, due to high interobserver variability, this criterion is strongly discouraged in the 2016 WHO classification. The vast majority of diffuse gliomas with histologic features of both astrocytic and oligodendroglial components can now be confidently classified as purely astrocytoma or oligodendroglioma based on genetic testing.
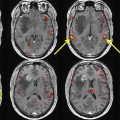
On imaging, oligodendrogliomas occur predominantly supratentorially, with preference for the frontal lobes (50%–65%). These tumors are relatively well defined, cortically based T2/FLAIR hyperintense masses with associated gyral expansion, occasional remodeling of the overlying calvarium, and frequent intratumoral coarse calcifications ( Fig. 5 ). Preoperative calcification was found significantly predictive of the 1p/19q codeletion. Peritumoral edema, hemorrhage, and necrosis are less common features. Similar to diffusely infiltrating astrocytomas, enhancement is inconsistent, with approximately 50% of cases manifesting some degree of enhancement, which is also unreliable in distinguishing grade II from grade III tumors.

Diffuse midline glioma, H3 K27M-mutant
Diffuse midline gliomas, H3 K27M-mutant, are a new grade IV entity in the 2016 WHO classification. Although these tumors are classically seen in pediatric patients, recent studies demonstrate that they are not infrequently encountered in the adult population. A recent case series of such tumors in adults reported the age range from ages 28 years to 81 years, with a median age of 52 years. Prognosis in the adult population is poor, similar to that in the pediatric population, with mean survival of 9.3 months.
Typically found in the thalamus, pons, or hypothalamus, these tumors present as expansile T2/FLAIR hyperintense masses with variable enhancement ( Fig. 6 ). Interestingly, 2 of the 4 criteria for pathologic diagnosis of this tumor are based on its imaging appearance. The tumors must appear diffuse and midline in addition to supporting the histologic diagnosis of glioma with the genetic mutation of H3 K27M. Leptomeningeal dissemination has been reported.