Fig. 1
The UspDBD/EcRDBD heterocomplex bound to an idealized consensus IR-1 (Devarakonda et al. 2003) (a) and to the natural hsp27 pal (Jakób et al. 2007) (b). The structure of the UspDBD/EcRDBD heterocomplex formed with the natural quasi-palindromic EcRE (hsp27 pal ) (b) exhibits significant differences in comparison to the UspDBD/EcRDBD-IR1 structure (a). A comparative analysis clearly showed how the EcRDBD accommodates DNA-induced structural changes. In particular, a fragment of the C-terminal extension (CTE) (red circle) of the EcRDBD folded into an α-helix, which was not seen in the structure obtained with the IR-1 element. The structure of the UspDBD/EcRDBD-hsp27 pal (Jakób et al. 2007) confirms the biochemical studies revealing the importance of key residues and segments from the UspDBD and EcRDBD molecules that are involved in the specific recognition of the natural hsp27 pal (Niedziela-Majka et al. 2000; Grad et al. 2001; Orłowski et al. 2004) . The UspDBD/EcRDBD-IR-1 structure is based on the file deposited in PDB (accession code: 1R0O) (a) and the UspDBD/EcRDBD-hsp27 pal heterocomplex structure is based on the file deposited in PDB (accession code: 2HAN) (b). The structures were visualized with PyMOL 0.99 (Delano 2002)
The intrinsic plasticity and adaptability of the EcRDBD molecule may be a key element which enables the protein to recognize a broad range of EcREs. Although there have been in-depth studies on the structure and function of the ecdysteroid receptor in arthropods, the high-resolution structure of the full-length EcR/Usp heterodimer alone, in complex with a specific EcRE, or with partners crucial to 20E and JH action, is still waiting to be resolved.
2.2 Conformational Changes of the Ecdysteroid Receptor on the Natural Response Element-Fluorescence Studies
2.2.1 DNA Binding Domain Dynamics and Detecting the Bending of the hsp27 Response Element
Many years ago, transcription factors, including NRs, were shown to induce substantial distortions in DNA structure , which may have affected the transcription-inducing activity of other complexes (Shulemovich et al. 1995; Lane et al. 1992; Potthoff et al. 1996; Prendergast et al. 1996; Petz et al. 1997; Heyduk et al. 1997) . As described above, the UspDBD preferentially binds the 5’half-site of hsp27 pal and acts as an anchor that dictates the polarity of the EcR/Usp heterocomplex (5’-Usp-DBD-EcR-DBD-3’) (Niedziela-Majka et al. 2000) . The final structure depends on both protein conformation and a regulatory element. The UspDBD alone and the UspDBD/EcRDBD heterodimer induce hsp27 pal bending, however, the UspDBD plays a crucial role in defining the overall architecture of the UspDBD/EcRDBD-hsp27 pal complex (Niedziela-Majka et al. 2000; Dobryszycki et al. 2006) . In contrast to the UspDBD, the EcRDBD is flexible and it causes a slight additional conformational change in the preformed structure (Dobryszycki et al. 2006; Orłowski et al. 2004) . Nevertheless, FRET analysis (fluorescence resonance energy transfer) indicated that both proteins are distorted, which changes the distances between the respective protein ends and the resulting DNA ends (Fig. 2). These spatial relationships indicate that when the UspDBD bound to the regulatory element 5’half-site there is a reduction in the distance between the ends of hsp27 pal and bending of about 23o (Dobryszycki et al. 2006; Jakób et al. 2007) . The binding of UspDBD brings both ends of the DNA to the C-terminus of the UspDBD. This may be the result of DNA bending, but it is also most likely the result of a conformational change in the protein (Fig. 2a). The binding of the EcRDBD to the DNA 3’half-site, in the presence of the UspDBD at the 5’half-site, does not significantly alter the DNA bending angle, but it clearly causes the C-end of the UspDBD to be about 6Å from both the 5’- and 3’-ends of hsp27 pal . This indicates that there is a strong conformational change in the UspDBD (Fig. 2b). The changes in the distances could have come from structural distortions in the protein or the regulatory element. An open question remains concerning DNA hinge point(s) and the best model to use to describe bending. It is important to note that the FRET results were in full agreement with the X-ray structure.
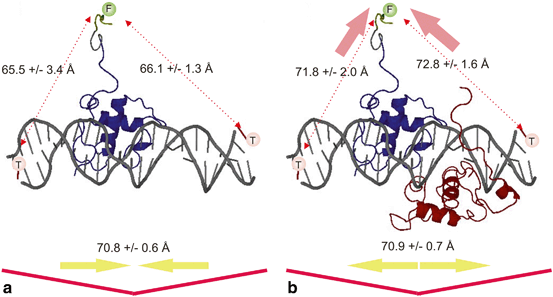
Fig. 2
Changes in the distances in the complexes between the UspDBD (blue ribbon) and the hsp-27 pal regulatory element in the absence a and in the presence b of the EcRDBD (brown ribbon). The figure was prepared on the basis of the structure of the complex of the ecdysteroid receptor DBDs with hsp27 pal obtained from X-ray experiments (Jakób et al. 2007). Taken from Pakuła et al. (2012) (Pakuła et al. 2012)
DHR38 is the Drosophila ortholog of the mammalian NGFI-B subfamily of orphan NR, which includes NGFI-B (NR4A1), Nurr1 (NR4A2), and NOR1 (NR4A3). Although members of the NGFI-B family are thought to function exclusively as monomers, DHR38 in fact interacts strongly with Usp, and this interaction is evolutionarily conserved (Sutherland et al. 1995; Dutko-Gwóźdź et al. 2008) . DHR38 can repress transcription of a reporter gene under the control of the hsp27 pal . In vitro experiments have shown that full-length DHR38 can form stable complexes with Usp, and thus can compete with EcR as a dimerization partner for Usp, which consequently disrupts the binding of EcR/Usp to the response element (Sutherland et al. 1995) . Surprisingly, recent data indicated that the EcRDBD is able to interact with the DHR38DBD on the hsp27 pal (Zoglowek et al. 2012) . Based on the end-to-end distances between donor and acceptor probes, hsp27 pal bend angles were estimated using FRET method (Dobryszycki et al. 2006) . The results obtained from FRET measurements revealed that there had been shortening of the end-to-end distance of hsp27 pal, both in the presence of the EcRDBD and also in the presence of the EcRDBD and DHR38DBD together. This corresponds to the respective DNA bend angles of about 36.2 ° and 33.6 °, respectively. The small 3.4 ° decrease in the bend angle in the presence of both proteins suggests that the key factor involved in the bending is the binding of the EcRDBD molecule to the 5’-regulatory element half-site. It seems that the EcRDBD induced a conformational change in hsp27 pal and created the conditions needed for DHR38DBD binding. It is worth noting that the interaction of the EcRDBD induced bending in hsp27 pal , while subsequent binding of the DHR38DBD did not significantly change the overall distortion of the regulatory element.
2.2.2 Cooperation Within the Protein-Protein Interaction Between the UspDBD and EcRDBD
Quantification of the level of DNA-binding activity of specific proteins is one of the most commonly performed experiments in biomedical research. Molecular beacons (MBs) use FRET measurements to investigate ssDNA hybridization and protein–DNA interactions in solution (Dummitt and Chang 2006; Li 2000; Fang et al. 2000; Tyagi and Kramer 1996; Heyduk et al. 2003; Heyduk and Heyduk 2002; Knoll and Heyduk 2004; Krusiński et al. 2008) . A technology based on MBs also makes it possible to quantitatively investigate proteins that interact with DNA (Krusiński et al. 2010). The double molecular beacon (DMBs) system has been developed for independent quantitative analysis of the binding affinity of dimerizing DNA-binding proteins with two specific DNA sites in proximity to each other. The use of DMBs showed that EcRDBD/UspDBD interactions mediate the cooperative binding of the ecdysteroid receptor DBDs to hsp27 pal . An analysis of the microscopic dissociation constants obtained with a DMB led to the conclusion that there is increased affinity of the UspDBD to the 5’half-site in the presence of the EcRDBD when the 3’half-site was occupied, and there was increased affinity of the EcRDBD to the 3’half-site when the 5’half-site was occupied. This cooperative effect was quantified by the respective dissociation constants, clearly indicating that protein-DNA interactions are strongly influenced by UspDBD/EcRDBD interactions.
2.3 The EcRLBD/UspLBD Heterocomplex
To date, five crystallographic structures have been described for the LBDs of EcR/Usp heterodimers with bound ligands. The reported structures are from moth Heliothis virescens (Lepidoptera) (Browning et al. 2007) the whitefly Bemisia tabaci (Hemiptera) (Carmichael et al. 2005) and the beetle Tribolium castaneum (Coleoptera) (Iwema et al. 2007) in complex with phyto-ecdysteroid ponasterone A (ponA), which differs from 20E only in the lack of the 25-hydroxyl group. LBDs from Heliothis was also reported in complex with 20E (Billas et al. 2003) . There is also an additional structure that was reported from Heliothis EcRLBD/UspLBD in complex with a non-steroidal, lepidopteran-specific agonist BYI06830 dibenzoylhydrazine (DBH insecticide) (Billas et al. 2003). The overall architecture of the EcRLBD and UspLBD that makes up each heterodimer is similar to that seen in the crystal structures of other NR LBDs, with a general fold consisting of a three-layered, antiparallel, α-helical sandwich and a β-sheet. The ligand-binding pocket has a J-shaped architecture that extends from helix H12 to helix H5 and the β-sheet, and it is completely buried inside the receptor. The overall structure of the Heliothis heterodimer EcRLBD/UspLBD bound to 20E (Fig. 3) is highly homologous to the structure of the ponA bound Heliothis, Bemisia, and Tribolium receptor complexes; the interactions between the residues of the ligand-binding pocket and 20E or ponA are highly conserved (Browning et al. 2007) . In their respective ligand bound EcRLBD/UspLBD heterodimer crystal structures, 20E and ponA adopt almost identical chair conformations and have similar positioning of their alkyl tail (Browning et al. 2007). The ligand/receptor interactions are similar, except for the extra hydrogen bond created between the 25-OH group of 20E and the polar residue Asn504 in helix H11 (Browning et al. 2007). Remarkably, the binding of two structurally and chemically distinct ligands, ponA and DBH, resulted in Heliothis EcR adopting a different conformation. In particular, the corresponding ligand-binding pockets were found to only partially overlapped and displayed different sizes and shapes (Billas et al. 2003) . For EcRLBD structures, the β-sheet forming ligand-binding pocket is composed of three strands. This is a key region of the receptor that displays unusual flexibility in response to different ligand types. For synthetic DBH agonists , a complete rearrangement of the region of the β-sheet was observed, involving two aromatic residues, Phe397 and Tyr403, that changed conformation to fill the space left unoccupied by the small DBH molecule (Billas et al. 2003). Another critical region is the connection between helices H1–H3, which in 20E- and ponA-bound EcR structures features a small helix H2 that is absent in the complex with the DBH compound (Billas et al. 2003). The existence of this small helix seems to be closely linked to the existence of direct and indirect stabilizing interactions with the ligand. Because the small DBH molecule is not located in close vicinity to the region comprising helix H2 and the β-sheet, H2 is not ligand-mediated and unfolds into a loop (Billas et al. 2003; Billas and Moras 2005). All this crystallographic data indicate that the ligand binding domain of EcR is characterized by unusual high degree of flexibility. As mentioned above, EcRDBD also displays flexibility and adaptability. The EcRLBD/UspLBD heterodimer interface is comprised of an intricate network of hydrophobic and polar interactions mediated in both partners by the helices H7, H9, H10 and the loop connecting helices H8 and H9 (L8-9). The interface of the Heliothis EcRLBD/UspLBD differs from that of the Bemisia EcRLBD/UspLBD or the Tribolium EcRLBD/UspLBD. In the Heliothis EcRLBD/UspLBD, the heterodimeric arrangement is more compact than in the Bemisia and Tribolium EcRLBD/UspLBD, due to the concomitant movement of structural elements from both partners and an additional zone that contributes to the heterodimerization interface involving of EcR H7 and Usp L8-9 (Iwema et al. 2007) . The dimerization interface observed for Bemisia and Tribolium EcRLBD/UspLBD is similar to interface observed for RXR in vertebrate NR heterodimers, where RXR L8-9 does not contribute to heterodimer contacts (Iwema et al. 2009),
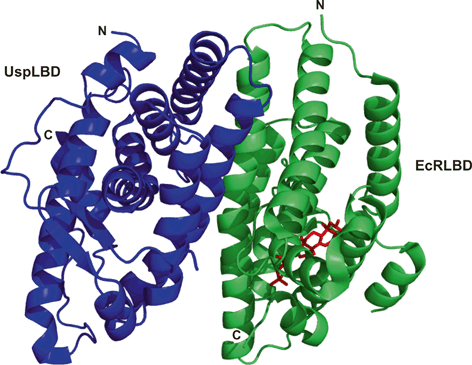
Fig. 3
The structure of Heliothis Usp in the complex (Browning et al. 2007) is almost identical to the structure of the monomer (Billas et al. 2001) , with a large hydrophobic ligand-binding pocket that is filled with a phospholipid molecule. A similar X-ray structure was also observed for the Drosophila UspLBD (Clayton et al. 2001) . These structures are significantly different in comparison to human RXRLBD structures (Egea et al. 2000), the activation helix H12 is locked in a so-called antagonist conformation by intra-protein interactions with a structural motif conserved in Mecopterida Usps (Clayton et al. 2001; Browning et al. 2007). A structure without a ligand-binding pocket was reported for the Bemisia UspLBD (Carmichael et al. 2005) and the Tribolium UspLBD (Iwema et al. 2007). The LBPs are filled by residues that stabilize an apo conformation and H12 is positioned in a so-called antagonist conformation, (Carmichael et al. 2005; Iwema et al. 2007) . The structure of the ligand-binding pocket of Usp was highly plastic during insect evolution, adopting three of the four states known to NRs: a nutritional sensor (basal insects), a real orphan (Hemiptera, Coleoptera) and a receptor with constitutive activity (Mecopterida) (Chaumot et al. 2012) . Maintenance of the ecdysone pathway was achieved, at least in part, through the molecular adaptation of the UspLBD (Iwema et al. 2007) . Some experimental data suggest that JH is the Usp ligand (Jones and Sharp 1997; Xu et al. 2002) .
3 Structural Analysis of Disordered Regions
In addition to the highly evolutionarily conserved, globular DBD and LBD, which have a stable fold, there are also IDRs, i.e. the A/B region called the NTD which is hypervarible in sequence and length, the D hinge region linked to the DBD and F region which only a few NRs possess (McEwan 2009) . IDRs and intrinsically disordered proteins (IDPs) are characterized by the lack of a stable and unique three-dimensional structure under physiological conditions. They are able to adopt distinct structural features upon interaction with particular protein partners or small molecular ligands (Uversky 2002; Wright and Dyson 1999) . Additionally, their binding functions can be altered by posttranslational modifications (PTMs) (Uversky et al. 2008). Structural intrinsic disorder (ID) is thought to be essential to these proteins, as their various biological functions stem either directly from this state or from some local folding or ordering during molecular recognition (Tompa 2002) . Comprehensive bioinformatic analysis provided evidences of ID in NR families (Krasowski et al. 2008) . The ID content of nearly 400 NRs in particular regions or domains was calculated across the full sequence. The NTD and hinge region was found to have a nearly 2.7 times higher probability of ID than the LBD across all species.
Despite the fact that there have been extensive studies on the structure and function of the arthropod functional ecdysteroid receptor and its partners in 20E signalling, there have been only three reports detailing the molecular characteristics of the NTDs isolated from insects: the EcRNTD (Nocula-Ługowska et al. 2009) and the DHR38NTD from Drosophila (Dziedzic-Letka et al. 2011) and the UspNTD from Aedes aegypti (Pieprzyk et al. 2014) . There is only one report on the Drosophila EcR hinge region (Zoglowek et al. 2012) and nothing is known about the F region. One of the three reports thoroughly describes the structural properties of the Drosophila EcRNTDs (Nocula-Ługowska et al. 2009) . In Drosophila there are three EcR isoforms, EcRA, EcRB1, and EcRB2 that exhibit diverse spatial and temporal distributions within various tissues and reveal important functional differences (Talbot et al. 1993) . Recently the structural properties of the NTDs of EcRA and EcRB1 isoforms have been reported. In silico analysis performed using different bioinformatic tools showed the existence of large IDRs in both NTDs. Moreover, analysis done with various techniques, e.g. CD spectroscopy or size-exclusion chromatography showed that both NTDs had features of the collapsed, disordered conformation that resembles that of pre-molten (PMG)-like IDPs (Nocula-Ługowska et al. 2009) . However, the analysed NTDs have distinct structural properties. In particular, the Drosophila EcRANTD has a lower content of a regular secondary structure than the EcRB1NTD and the EcRANTD is less compact than the EcRB1NTD. It has been previously reported that these isoforms reveal different transcription activities depending on the promoter used and the type of host cells (Dela Cruz et al. 2000; Hu et al. 2003; Mouillet et al. 2001) . It has been postulated that the functional differences reported for the EcRA and EcRB1 isoforms might be the result of subtle structural differences that were observed in their NTDs (Nocula-Ługowska et al. 2009). Dziedzic-Letka et al. (Dziedzic-Letka et al. 2011) also studied the structural characteristics of the NTD, but from another member of the NR family from Drosophila, DHR38 (Dziedzic-Letka et al. 2011). Studies on the function of DHR38 indicated that this NR is involved in 20E (Baker et al. 2003; Sutherland et al. 1995) . It was suggested that the DHR38NTD may be involved in some protein-protein interactions that are critical for the transmission of an ecdysteroid-dependent signalling pathway (Dziedzic-Letka et al. 2011). Despite the fact that the DHR38NTD has an amino acid composition that is different from that of the EcRNTDs, both, bioinformatics analysis and comprehensive biophysical and biochemical analyses showed that the DHR38NTD exhibited characteristics reminiscent of a PMG-like IDP with a partially unfolded conformation and regions of secondary structures, similarly as was observed for the EcRNTD. Moreover, size-exclusion chromatography in denaturing conditions and CD spectroscopy in the presence of osmolytes, which are known to promote a function-related local structure, showed that the DHR38NTD could adopt a disordered state or more ordered conformations in response to changes in environmental conditions. It was suggested that the structure of the DHR38NTD could have been influenced by multiple phosphorylation that promoted and mediated distinct signalling effects. Because of the formation of different conformational states, many distinct interaction surfaces might be formed and recognized by specific protein partners (Dziedzic-Letka et al. 2011) . Further detailed studies are necessary to see if phosphorylation can indeed induce conformational changes in the DHR38NTD. Recently, increasingly more attention has been paid to UspNTD. In the mosquito Aedes aegypti two Usp isoforms, UspA and UspB, have been cloned and identified (Kapitskaya et al. 1996; Wang et al. 2000) . Recently biochemical and biophysical properties of the UspBNTD have been described (Pieprzyk et al. 2014) . The results showed that the UspBNTD is an IDP which has residual secondary structures. The anomalous behaviour of the UspBNTD in SDS-PAGE electrophoresis and in secondary structure analyses by far-UV CD spectroscopy and size exclusion chromatography indicated features of an IDP. However, in contrast to the Drosophila EcRANTD, EcRB1NTD and DHR38NTD which were reported to have been PMG-like IDPs, the Aedes UspBNTD cannot be unequivocally classified into one of the recognized classes of IDPs, based on its amino acid composition, molecular mass, Stokes radius and secondary structure content. Nevertheless, the sedimentation velocity analytical ultracentrifugation experiment determined that Aedes UspBNTD had the shape of an asymmetric, elongated and elliptically-shaped protein. Similarly to the NTDs described above, the UspBNTD has the potential to form structures in the presence of a folding/refolding agent. This again proves the assumption that these NTDs have the ability to exist in a wide range of conformational states and may have the ability to bind to several different partner proteins, thus exhibiting multiple functions as with other IDPs (Wright and Dyson 1999) . What is particularly noteworthy and what makes the Aedes UspBNTD radically different from other insect NTDs that have been described is the fact that the UspBNTD exhibited a tendency for homooligomerization and probably coexists in solution as a monomeric and dimeric species (Pieprzyk et al. 2014) . A comparison was made of the propensity for oligomeric forms in other insect NTDs, namely the NTD of Usp from Drosophila and the NTD of Usp from Bombyx mori. The dimerization potential is conserved among these receptors, but to a different extent within each species. The Aedes UspBNTD exhibits the highest tendency to form a dimer, then the homologs from Drosophila and Bombyx. Rymarczyk et al. (Rymarczyk et al. 2003) suggested that the Drosophila UspNTD probably plays an important role in stabilizing the receptor oligomers in solution and may possess an additional interface for dimer interaction or may be a part of existing interfaces within the full-length protein (Rymarczyk et al. 2003). Pieprzyk et al. (Pieprzyk et al. 2014) suggested that the NTDs from Aedes, Drosophila and Bombyx may have different structural architecture and that the specifics that determine dimerization might be distributed differently within each domain (Pieprzyk et al. 2014) . Despite differences in the amino acid sequences (Fig. 4), all the insect NTDs described above exhibit properties of IDP molecules and have a pliable structure. They were not completely random coil-like, since there was content of a residual secondary structure. It is worth emphasizing that although they were all classified as IDRs, there are subtle differences in their structure, like a different degree of secondary structure or compactness or differences in hydrodynamic properties any of which may result in different activity (Pieprzyk et al. 2014). Moreover, the NTDs may be involved in dynamic protein-protein interactions through their ability to adopt distinct conformations. They may also regulate mechanisms in signal transduction, e.g. by having different tendencies for dimerization. IDPs and IDRs, while structurally poor, are functionally rich by virtue of their flexibility and modularity (Malaney et al. 2013) . There is evidence indicating an intimate relationship between PTMs and structural disorder (Gao and Xu 2012) . These modifications involve low affinity, high specificity binding interactions between a specific enzyme and the protein that is modified (Xie et al. 2007) . Based on observed correlations between PTMs and predicted disorder, it was shown e.g. that IDPs/IDRs are substrates of twice as many kinases as ordered proteins are (Uversky et al. 2008) . By comparing a collection of more than 1500 experimentally determined S, T, and Y phosphorylation sites to potential sites that were nonphosphorylated, the segments surrounding phosphorylation sites were found to be significantly enriched in amino acids that are usually in IDRs (Iakoucheva et al. 2004) . The majority of kinases, whose substrates were mostly IDPs/IDRs, were either regulated in a manner that is dependent on the stage of the cell division cycle or were activated upon exposure to particular stimuli or stress. Therefore, protein modifications may not only serve as important regulatory mechanisms that fine-tune the functions of IDPs or IDRs, but they may also be necessary to tightly control the availability of these proteins under different conditions (Uversky et al. 2008) .
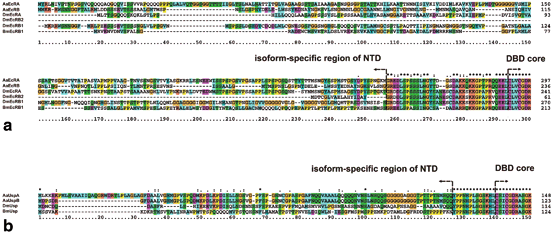
Fig. 4
Alignment of the NTD sequences of selected insect species. a Amino acid alignment of the N-terminal domain of EcR from Aedes aegypti (UniProtKB: P49880), Drosophila melanogaster (P34021), Bombyx mori (P49881); b Amino acid alignment of the N-terminal domain of Usp from A. aegypti (Q9GSG8; UspBNTD: Q9GSG7), D. melanogaster (P20153), B. mori (H9J9J0). The dots indicate conservation between groups of weakly similar properties; colons indicate conservation between groups of strongly similar properties and asterisks indicate positions which have a single, fully conserved residue. All alignments were done using ClustalX (Thompson et al. 1997)
Relatively little work has focused on the PTMs of insect NRs. Phosphorylation was shown to play an important role in regulating the function of the EcR/USP complex (Sun and Song 2006) . Both EcR and Usp are phosphoproteins and their phosphorylation is regulated by 20E (Song and Gilbert 1998; Rauch et al. 1998; Nicolai et al. 2000) . However, only the site of modification in Drosophila Usp was identified. Using liquid chromatography-tandem mass spectrometry, the site of protein kinase C (PKC) phosphorylation was found to be located in the NTD (S35) (Wang et al. 2012) . BLAST search against the NCBI protein database revealed that S35 of Usp is well conserved in other Diptera. However, there is no information about how this modification impacts the structure of the NTDs. Additionally, nothing is known about the sites of EcR phosphorylation. Bioinformatics tools indicated that putative phosphorylation sites are present mainly in the NTD (Rauch et al. 1998), but this requires experimental verification.
Sumoylation is deeply involved in PTMs and has a significant influence on the structure of NR NTDs. This is a protein conjugation resembling ubiquitination, based on the reversible attachment of the small ubiquitin-related modifier (SUMO) protein (Geiss-Friedlander and Melchior 2007) . At least some insect NRs have the SUMO peptide added to a K residue in their NTDs. Drosophila EcR undergoes isoform-specific multi-sumoylation. The pattern of modification remains unchanged in the presence of the ligand and the dimerization partner. The SUMO acceptor sites are located in the NTD, DBD, LBD and region F. However, the most interesting modification site is in the NTD as it encompasses only isoform A (Seliga et al. 2013) . This was also in agreement with what had been predicted with bioinformatics tools (Watanabe et al. 2010) . A comprehensive structural comparison revealed that the A isoform-specific region of EcR from different species of arthropods contained evolutionally conserved microdomain structures including the sumoylation motif. Sumoylation of the A isoform-specific region might contribute to transcriptional regulation that is either cell-type or organism dependent and mediated by the NTD of the EcR A isoform (Watanabe et al. 2010). The consequences of modification and the resulting impact on conformation and function may be especially crucial for the disordered sequences in these areas. Sumoylation may influence the structure and mode of action both EcR and its partner, Usp. The main sites of sumoylation of Drosophila Usp are located in the NTD (Bielska et al. 2012) , where three K residues can be sumoylated, but the attachment of one SUMO molecule prevents modification of the other two remaining sites. One possible explanation for this phenomenon could be steric hindrance, as all K residues in the NTD lie close to each other and the attachment of one SUMO molecule can apparently block the modification of others. However, monosumoylation of the NTD is most likely caused by interaction with proteins which can lock the NTD region in a specific conformation, exposing one of the three K residues to SUMO modification. Alternatively, monosumoylation might be a consequence of conformational changes after SUMO modification. It has been suggested that the sumoylation of Usp could be an important factor that modulates its activity by changing inter- and intra-molecular interactions (Bielska et al. 2012). Interestingly, the amino acid sequence alignment of the NTDs of Usp from Diptera, Hymenoptera, Coleoptera and Hemiptera showed that the K20 residue identified as a SUMO acceptor site and the embedded sequence in Drosophila Usp in the NTD is highly conserved in other classes of insects. This suggests that the K20 residue constitutes an important regulatory element of the transcriptional activity of Usp receptors (Bielska et al. 2012) .
Another IDR identified in NRs is the C-terminus of the LBD called the F region, which was mentioned above. This region has few structural features, displays little evolutionary conservation and reveals no clues as to the function of the sequence (Hu et al. 2003) . It had been suggested that the F region might play a role in recruiting a co-activator in the E domain and in determining the specifics of the LBD co-activator interference (Laudet 2002) . The F region of EcR from different species consists largely of repeated residues, and there is no apparent conservation of its sequence. Drosophila EcR contains an unusually long F region (223 residues), for which there is no known function. By contrast the C-terminal region in EcRs from other flies are significantly shorter—most NRs contain an F region of 10–50 residues (Hu et al. 2003). The F region of insect NRs, similarly to the NTD, is also a target of PTMs. The K residue in the F region of each Drosophila EcR isoform (K871, K842 and K662, respectively for EcRB1, EcRA and EcRB2) was shown to be subjected to sumoylation (Seliga et al. 2013) . Due to the variability in length and sequence among EcRs, it is difficult to assume that EcRs from species other than Drosophila might be sumoylated in F region.
Drosophila Usp was also shown to be modified by SUMO in the F region (K506) (Bielska et al. 2012) . The F region was analyzed together with the E region, because the F region has only 12 aa and is too short to be independently investigated. In the isolated E/F region of Usp there are two alternatively sumoylated residues: K424 (E region) and K506 (F region). However, these residues are not available for sumoylation in the full-length protein. The main sites of sumoylation of full-length Usp are located in the NTD, although sumoylation of some residues in E or F regions is also possible (Bielska et al. 2012). This adds another level of complexity to NR signal integration, since PTMs might influence the properties of the encoded functions.
Related posts:
 Multi-Functional Androgen Receptor Coregulator and Proto-Oncogene Melanoma Antigen-A11 (MAGE-A11)
Multi-Functional Androgen Receptor Coregulator and Proto-Oncogene Melanoma Antigen-A11 (MAGE-A11)
 Estrogen Receptor Modulators (SERMs) and Selective Androgen Receptor Modulators (SARMs)
Estrogen Receptor Modulators (SERMs) and Selective Androgen Receptor Modulators (SARMs)
 and Regulation of Nuclear Receptor Corepressor Complexes
and Regulation of Nuclear Receptor Corepressor Complexes
 Receptor Structure and Function
Receptor Structure and Function
 Analysis of Heterodimeric Nuclear Receptors
Analysis of Heterodimeric Nuclear Receptors
 Regulation and Intrinsic Disorder in Nuclear Hormone Receptors
Regulation and Intrinsic Disorder in Nuclear Hormone Receptors
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


