Fig. 1
Steroidogenic pathways (both traditional and backdoor) responsible for the synthesis of androgens and estrogens from cholesterol. HSD-Hydroxysteroid dehydrogenase
All of the subsequent steps in androgen and estrogen synthesis are mediated by a class of enzymes called hydroxysteroid dehydrogenases (HSD). There are two classes of HSDs, namely 3β- and 17β-HSDs, depending on the carbon on which they perform the dehydrogenase activity. These enzymes are involved in the oxidation and reduction of ketone and β-hydroxyl groups at the C3 and C17 positions of androgen and estrogen precursors. The major biological functions of these 17β-HSDs include both biosynthetic activation and inactivation of various estrogens and androgens. Fourteen mammalian 17β-HSDs have been identified to date; and are grouped into oxidative enzymes (17β-HSD types 2, 4, 6, 8, 9, 10, 11, and 14) that catalyze the NAD+-dependent inactivation of sex hormones and reductive enzymes (17β-HSD types 1, 3, 5, and 7) that catalyze the formation of more potent steroid receptor ligands. The proliferative effects of androgens and estrogens in target tissues and over-expression of 17β-HSDs in cancer have led to intense drug discovery efforts to identify and develop isoform selective 17β-HSD inhibitors that can be used for the treatment of breast, prostate and endometrial cancers, neurological disorders, endometriosis, acne, hirsutism and several other hormone dependent and independent diseases. The final conversion of inactive or less active androgens to active androgens, testosterone and DHT, is mediated by 17β-HSDs and 5α-reductase, respectively. Similarly, conversion of the weak estrogen, estrone, to estradiol is mediated by 17β-HSDs and conversion of testosterone to estradiol is mediated by the enzyme aromatase. Further inactivation of these active androgens and estrogens is mediated by various 17β-HSDs and 3α-HSDs.
The last 5 years have witnessed the discovery of “backdoor pathways” that synthesize active androgens and bypass the conventional routes of biosynthesis (Sharifi and Auchus 2012) (Fig. 1). Although these backdoor pathways make androgens in unconventional ways, they ultimately utilize the same set of enzymes used in traditional steroid biosynthesis in the testes. Estrogen biosynthetic pathways remain unchanged from conventional methods.
2.2 Tissues and Organs Involved in Androgen and Estrogen Biosynthesis
In addition to gonadal synthesis, local steroid biosynthesis produces testosterone and estradiol for the function of the many tissues. Estradiol synthesis in women of reproductive age primarily takes place in the granulosa cells of the ovary, mainly via conversion from estrone. In men, 15 % of circulating estrogens are synthesized in testes. However, recent evidence suggests that, in post-menopausal women and men undergoing andropause, adipose tissue and skin express steroidogenic enzymes that contribute significantly to local synthesis of these pivotal hormones. Aromatization of testosterone to estradiol plays important roles in estradiol synthesis in peripheral tissues including adipose, skin, brain, bone and others. In post-menopausal women and men, local expression of aromatase is important for synthesis of estradiol (Nelson and Bulun 2001). In post-menopausal women with osteoporosis and dementia, the expression of aromatase is lower in bone and brain, respectively, than that observed in healthy post-menopausal women (Simpson et al. 1997; Jakob et al. 1997; Bulun et al. 1999). Moreover, it was also demonstrated that local expression of aromatase increases with age and obesity, indicating that aromatization becomes primary estradiol synthesis machinery.
Androgen biosynthesis occurs predominantly in the testes. Preclinical studies and human trials indicate that castration leads to over 95 % depletion of circulating testosterone (Labrie 2011). The remaining approximately 5 % of androgens, which include DHT, testosterone, dehydroepiandrosterone (DHEA), DHEAS, androstenedione, androstenediol, and others, evolve from the adrenals. This finding resulted in the development of treatments (e.g., ketoconazole and abiraterone) for castration-resistant prostate cancers (CRPCs) that block adrenal androgen synthesis. In addition, negligible, but functionally relevant, androgen synthesis also takes place in the peripheral extra-gonadal tissues such as adipose.
2.3 Importance of Estrogens in Development, Physiology, and Pathology
Estrogens are also important for the development and physiology of various tissues. The role of estrogens is mediated by two receptors, ER-α (NR3A1) and ER-β (NR3A2). While ER-α is abundantly found in reproductive tissues, ER-β is expressed widely. Knock-out studies of the ER isoforms and aromatase helped identify the importance of estrogens in variety of physiological functions. These studies also demonstrated the overlapping and distinct functions of the two isoforms in mediating the actions of estrogens. While ER-α is considered a proliferative signal, ER-β is typically considered a repressor of ER-α action and an anti-proliferative isoform. ER-αKO mice were sterile, had high gonadotrophin production, lacked ovulation, had pubertal and mammary development abnormalities, osteoporosis, and estrogen insensitivity in the uterus. On the other hand, ER-βKO mice phenotypes were mild to moderate and did not or only mildly exhibited many of the above functional abnormalities (Emmen and Korach 2003; Hewitt and Korach 2003). On the other hand, functional abnormalities of ER-βKO mice were exacerbated when challenged with external stimuli such as a high fat diet or inflammatory molecules including lipopolysaccharide (LPS). In addition to these physiological functions, estrogens also play a pivotal role in many diseases, including cancers of the reproductive organs and cardiovascular disease. The physiological and pathological roles of estrogens are summarized in Table 1.
Table 1
Role of estrogens and androgens
Physiology | Pathology |
|---|---|
Estrogens | |
Secondary sexual organ development | Endometriosis, Breast: ovary. endometrial, prostate cancers |
Bone development and maintenance | Osteoporosis |
Brain development and cognition | Alzheimer’s, dementia |
Glucose and adipose homeostasis | Cardiovascular diseases |
Gonadotropin secretion for ovulation | Female pseudohermaphrodit ism and male tall stature with unfused epiphyses |
Fusion of epiphysis in men | Gonadotropin feedback irregularity |
Androgens | |
Secondary sexual organ development | Prostate cancer, sexual dysfunction |
Bone development and maintenance | Osteoporosis |
Muscle generation and maintenance | Sarcopenia |
Brain development and cognition | Dementia, Alzheimer’s |
Glucose and adipose homeostasis | Cholesterol imbalance, Cardiovascular diseases |
Sperm maturation and sexual activity | Gonadotropin feedback irregularity |
2.4 Importance of Androgens in Development, Physiology, and Pathology
Similar to estrogens in females, androgens are important reproductive hormones in males (Table 1). Androgen insensitivity syndrome and andropause in humans provide unequivocal evidence of the importance of androgens in the development of sexual organs including the penis, scrotum, seminal vesicles, prostate, vas deferens, and epididymis (Zhou 2010). In addition, AR knockout mice exhibit osteoporosis, muscle wasting, insulin resistance and metabolic syndrome, and late onset obesity. Humans with longer poly glutamine repeats in the N-terminal domain of AR develop Kennedy’s disease or spinal bulbar muscular atrophy (SBMA), a rare neuromuscular degenerative disease and distinct form of androgen insensitivity syndrome that is characterized by muscle weakness and neuronal deformity.
3 Selective Estrogen Receptor Modulators (SERMs)
The phenotypic and physiological evidence from preclinical animal models and human trials show that ERs are a rational and highly valuable therapeutic target for many conditions. Estradiol was initially proposed as a treatment for post-menopausal osteoporosis and female secondary sexual growth deficiencies. In fact, conjugated estrogen products remain a viable treatment option for many post-menopausal symptoms associated with estrogen deficiency. Shortly after the discovery of ER, estrogen antagonists were suggested as treatments for breast cancer, uterine cancer, and endometrial hyperplasia. However, the ubiquitous expression of ER-α makes it difficult to distinguish between pharmacologic effects in target tissues and off-target effects when treating with estradiol or antagonists. The following scenarios describe the characteristics of an ideal SERM and the various pathophysiological conditions that require tissue-selective ER modulators;
Scenario 1
Post-menopausal women frequently suffer osteoporosis and bone fracture as a result of depleted levels of circulating estrogens. Treating post-menopausal women with estradiol or an ER-α agonist is a viable option therapeutically. However, treating post-menopausal women with estradiol will unfortunately result in ER agonist activity not only in bone but also in breast and uterus, leading to increased incidence of breast and uterine cancers. In this scenario, a small molecule that functions as an agonist in bone, but spares breast, uterus, ovaries and other tissues is considered ideal.
Scenario 2
Hormonal imbalance and other stimuli that hyper-activate ER can result in breast cancer. ER-positive breast cancers are treated with ER antagonists or aromatase inhibitors. However, treating post-menopausal or middle aged women with pure estrogen antagonists results not only in antagonistic activity in breast and regression of breast cancer, but also results in osteoporosis, due to impairment of ER function in bone. In this scenario, a small molecule that functions as an antagonist in breast, but spares bone, uterus, and ovaries is desirable.
Scenario 3
Angiotensin II-induced cardiac hypertrophy and fibrosis as a result of hypertension can be rescued by estradiol through its actions on ER-β (Pedram et al. 2010). However, administration of estradiol will activate not only ER-β and alleviate cardiac hypertrophy and fibrosis, but will also activate ER-α, which can result in breast and endometrial cancers. In this scenario, isoform-selective small molecule agonists that activate ER-β, but do not cross react with ER-α, are highly desirable.
From the above three scenarios, an ideal SERM should be tissue- and isoform- selective and provide therapeutic benefits without associated side effects. Table 2 summarizes the desirable properties of ER ligands in various tissues and the profile for the “Ideal SERM”.
Table 2
Estrogen receptor ligands
Target tissue | Properties |
|---|---|
Breast | Antagonist |
Endometrium | Antagonist |
Platelets | Antagonist |
Bone | Agonist |
Cholesterol and metabolism | Agonist |
Climacteric | Agonist |
Vagina | Agonist |
Adipose (obesity) | Isoform-selective ERβ agonist |
Cardiovascular | ERα antagonist or ERβ selective agonist |
CNS | Agonist |
Prostate | ERα antagonist or ERβ selective agonist |
3.1 Clomiphene
Clomiphene (Fig. 2), belonging to the triphenylethylene structural series, was the first SERM. Its discovery in 1960 preceded the discovery of tamoxifen, the most widely used SERM, by 2 years. Clomiphene is an ER antagonist and the primary choice for ovulatory dysfunction, including polycystic ovary syndrome (PCOS). Ovulatory dysfunction, such as PCOS, luteal phase deficiency and anovulation are commonly observed reproductive abnormalities in women. As an ER antagonist, clomiphene citrate suppresses the negative feedback inhibition of estradiol and increases secretion of follicle stimulating hormone (FSH), which in turn stimulates follicular development and results in an increase in ovulation rate. Some of the common side effects of clomiphene are hot flashes, abnormal uterine bleeding, nausea and/or vomiting. Although clomiphene increases ovulation by 80–90 % in women with PCOS, it increases pregnancy only by 25–30 %. Endometrial biopsies obtained from women treated with clomiphene indicated that it reduces the thickness of the endometrium, an effect that is more pronounced with clomiphene than other SERMs or aromatase inhibitors.
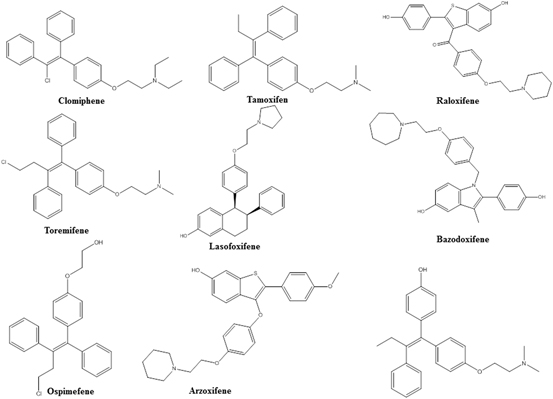
Fig. 2
Selective estrogen receptor modulators (SERMs)
Clomiphene citrate is also being investigated as a treatment for male hypogonadism. Utilizing the same phenomenon to inhibit the negative feedback loop of gonadotropins, hypo gonadal men treated with clomiphene demonstrated a significant increase in serum testosterone levels and in ADAM scores (Androgen Deficiency in Ageing Male). Interestingly, hypo gonadal men receiving clomiphene for more than 2 years also demonstrated an increase in bone mineral density (BMD) and a decrease in osteoporosis (Moskovic et al. 2012). Data on the changes in BMD in hypo gonadal men and women with PCOS by clomiphene is conflicting. While some studies indicate an increase in BMD with clomiphene, others show that clomiphene might decrease BMD. These seemingly contradictory results could be due to the direct antagonistic effects of clomiphene on ER-α in bone, indirect effects of clomiphene to increase testosterone and estrogens, or differences in the overall hormonal milieu of women and men.
Preclinical studies have indicated that while clomiphene is an antagonist in ovary and endometrium, it is an agonist in the skeletal and cardiovascular systems. Interestingly, while E-clomiphene and Z-chlomiphene both have agonistic activities in bone and cardiovascular tissues, E-chlomiphene is an agonist and Z-chlomiphene is an antagonist in uterus.
3.2 Tamoxifen
Tamoxifen (Fig. 2) was discovered in 1962 by Imperial Chemical Industry (now Astra Zeneca) as a triphenylethylene SERM. Tamoxifen is the most widely used SERM for breast cancer and it functions through its active metabolite, hydroxytamoxifen. Tamoxifen has become the most widely studied SERM from the mechanistic perspective as it functions as an ER antagonist in breast, but agonist in uterus and endometrium. Tamoxifen binds to ER with a high affinity of 2 nM and antagonizes both isoforms of ER.
Tamoxifen is currently in use for node-negative breast cancer in pre- and post-menopausal women and for node-positive breast cancer in post-menopausal women. It is also effective in metastatic breast cancer and has recently been approved for the prevention of breast cancer in high risk women. Multiple clinical trials have demonstrated significant survival benefits in women with breast cancer treated with tamoxifen (Davies et al. 2013; Darby et al. 2011). Being a SERM, prolonged use of tamoxifen provided beneficial anti-cancer effects in the breast and also modestly preserved BMD and decreased the number of fractures (Ryan et al. 1991; Love et al. 1992). Since post-menopausal women undergoing ER-antagonistic treatments have a higher incidence of osteoporosis and fractures, tamoxifen’s small, but beneficial, effects on bone are considered one of the advantages for improved survival of these patients. Recently, tamoxifen also received attention for its potential for favorable effects on cardiovascular diseases (Love et al. 1994). A number of cardiovascular diseases markers such as LDL, HDL, and triglycerides are favorably altered by tamoxifen, making it potentially more valuable for patients with breast cancer.
Despite its many beneficial effects, one major concern with the use of tamoxifen is its effects on the endometrium. Endometrial cancer is increased by 2–4 fold in randomized trials (Chen et al. 2014). Uterine-related symptoms include vaginal bleeding and leucorrhoea. Multiple lines of evidence suggest that while tamoxifen functions as an ER antagonist in breast, it functions as an agonist in the uterus and endometrium (Wood et al. 2010). This warrants careful monitoring of breast cancer patients receiving tamoxifen for uterine cancer.
3.3 Raloxifene
Raloxifene (Fig. 2) belonging to the benzothiophene structural series was discovered and developed by Eli Lilly for post-menopausal osteoporosis. Raloxifene provides unique SERM properties with one of the best profiles known. While raloxifene functions as an ER agonist in bone and in the regulation of serum lipids, it functions as an antagonist in breast and uterus, making it a highly valuable tool to combat post-menopausal osteoporosis without the disadvantages of activating ER in other tissues (D’Amelio and Isaia 2013). Studies on the effects of raloxifene on bone turnover markers indicated that raloxifene prevented bone loss and fractures at multiple sites with concurrent increase in BMD (D’Amelio and Isaia 2013). Bone turnover markers were significantly reduced and bone formations markers were up-regulated by raloxifene.
Recent evidence suggests that raloxifene significantly prevented breast cancer incidence in post-menopausal women, with results comparable to that of tamoxifen (Vogel et al. 2006). Women treated with raloxifene demonstrated a breast cancer incidence of only 1.7 per 1000, whereas placebo-treated women had a breast cancer incidence of 3.7 per 1000. The outcome of the Study of Tamoxifen and Raloxifene (STAR) trial indicated that raloxifene was as effective as tamoxifen in reducing invasive breast cancer without any thromboembolic side effects (Freedman et al. 2011). While raloxifene had a better benefit/risk ratio than tamoxifen in post-menopausal women with a uterus, raloxifene and tamoxifen had comparable benefit/risk ratios in post-menopausal women who had undergone hysterectomy. Overall, the clinical data indicates that raloxifene has a better benefit/risk ratio than tamoxifen.
Raloxifene lowered LDL cholesterol without affecting HDL cholesterol, suggesting that it might also have beneficial effects on these markers of cardiovascular disease (Francucci et al. 2005). Raloxifene also showed favorable effects on CNS function, including effects on climacteric symptoms such as hot flashes. Among all the commercially available first-generation SERMs, raloxifene is considered by many to have a near-perfect SERM profile, with favorable benefits in desired organs and a lack of unwarranted side effects.
3.4 Toremifene
Toremifene (Fig. 2) differs from tamoxifen by only one chlorine atom and possess properties comparable to that of tamoxifen (Buzdar and Hortobagyi 1998). Toremifene is also used for the treatment of advanced breast cancer. While toremifene functions as an ER antagonist in breast, it functions as an agonist with regard to bone, lipids, and uterus. Toremifene was also tested as a treatment for osteoporosis in men undergoing androgen deprivation therapy (ADT) (Smith et al. 2011). Although it protected bone loss in men undergoing ADT, the studies necessary to establish its efficacy and safety in this population were never completed. Similar to tamoxifen, toremifene’s active N-desmethyl and 4-hydroxylated metabolites bind to and antagonize ER more potently than the parent molecule.
3.5 Second Generation SERMs
Several SERMS such as lasofoxifene, arzoxifene, ospemifene, afimoxifene, arzoxifene, and bazodoxifene (Fig. 2) have been approved since 2000 (Komm and Chines 2012). All of these SERMs offer distinct tissue- selective properties and an unique profile compared to the first generation SERMs described above. While lasofoxifene has ER agonistic activity in bone, it has antagonistic activity in breast and uterus making it suitable for the treatment of osteoporosis, breast cancer, and vaginal atrophy. Phase III trials with lasofoxifene showed that it increased BMD significantly without any associated endometrial or thromboembolic effects (Malozowski 2010). In other trials, lasofoxifene improved vaginal atrophy, reduced ER-positive breast cancer growth and coronary disease risk (LaCroix et al. 2010; Ensrud et al. 2010; Goldstein et al. 2011). Similarly, bazodoxifene reduced the incidence of osteoporosis in post-menopausal women without causing an increase in endometrial hyperplasia (Miller et al. 2008). As second generation SERMs, these compounds have better safety profiles and exhibit near ideal SERM properties such as ER antagonistic effects in breast and uterus and agonistic properties in bone, blood and CNS, making them more broadly applicable to many women’s health issues.
3.6 ER-β Selective Ligands
The effects of estrogens are mediated by two isoforms of ER, ER-α and ER-β. Although the LBD of the two isoforms is only 59 % homologous, the ligand binding pocket (LBP) differs only by two amino acids, making them highly similar (Katzenellenbogen 2011). Despite this striking similarity, academic investigators and pharmaceutical companies have successfully developed ligands that bind preferentially to one or the other isoforms in order to take advantage of the beneficial effects of the individual isoforms without impinging on the function of the other isoform. Several structurally diverse molecules have been identified that bind to ER-β at low nanomolar to picomolar affinity, without cross reacting with ER-α (Fig. 3).
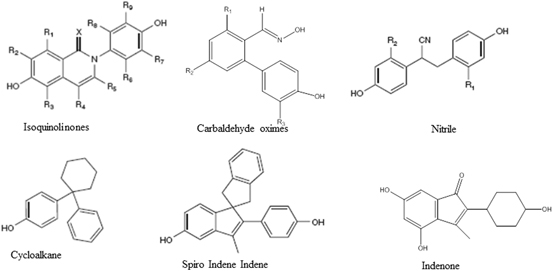
Fig. 3
Estrogen receptor β selective ligands
Diverse pharmacophores ranging from a simplistic linear diphenolic compound that achieves an inter-phenolic distance similar to estradiol to highly complex structures have been identified (Minutolo et al. 2011). The most common ER-β agonist template configuration is a (hetero) bicyclic compound substituted with a phenyl side chain such as genistein. Other templates include carbaldehyde oxime derivatives (Wyeth), hydroxyl-biphenyl carbaldehyde oximes (Wyeth), diaryl substituted salicylaldoximes (UIUC), alkyl linked (UIUC), cycloalkanes-linked biphenyls (Acadia), spiro indene-indenes (Merck) and many more. Academic and industrial groups have patented close to 50 different structural templates as ER-β ligands. These templates represent a heterogeneous mixture with regard to their level of characterization and degree of design success. Although several groups have been successful in discovering and developing high affinity ER-β-selective templates, Wyeth (now Pfizer) stands out as the leader in the field in terms of attaining extremely high affinity ligands and exploring a wide variety of templates. Further, Wyeth is the only pharmaceutical company to advance a synthetic ER-β agonist, prinaberel (ERB-041), to clinical trials. They explored a variety of clinical indications, including rheumatoid arthritis, Crohn’s disease, and endometriosis. However, the data were neither convincing nor published in entirety. The only other clinical candidate ER-β agonist, MF-101 (Bionovo) is an herbal extract containing a combination of ER-β agonists of plant origin. However their recently reported clinical trial results were only marginally successful in suppressing post-menopausal hot flashes. Diarylpropionitrile (DPN), which was initially developed by Katzenellenbogen et al., has been used extensively for academic exploration of the role of ER-β (Meyers et al. 2001).
ER-β agonists have been highly sought agents following the discovery of ER-β in 1996 (Kuiper et al. 1996). The distinct tissue-distribution alone seemed to suggest that tissue-selective estrogenicity was possible, which is further supported by the diverse and often opposing functional roles of ER-β, compared to ER-α. The theoretical problem of designing ER-β-selective agents was fully defined following the first crystal structure in 1999 of genistein bound to the ER-β LBD. Combined with the earlier ER-β LBD structures, this work revealed that the polar interactions available within the ER-α and ER-β ligand binding pockets are identical. Importantly, the ER-α and ER-β ligand binding pockets only varied by two amino acids, M336/L384 and I373/M421, producing topological and pocket size differences that necessarily must be the basis for subtype selectivity.
Multiple ER-β selective chemotypes with promising preclinical activity exist across a very broad chemical space. Many of these molecules have demonstrated promise as potential treatments for inflammation (Wyeth), cardiovascular diseases, obesity, and metabolic diseases (GTx), hormone therapy (Wyeth and Bionovo for endometriosis and post-menopausal symptoms) and prostatic disease (Eli Lilly). However, only a small fraction of the theoretically useful indications have been explored clinically, leaving room for exploration of many clinical indications including anxiety or depression, colon cancer, hepatic fibrosis, memory, neuroprotection, neuropathic or inflammatory pain, and wound healing, to name just a few.
Further clinical testing is needed to better understand the beneficial effects and liabilities of ER-β agonists in man. Nonetheless, the breadth and depth of ER-β agonists suggest that the preclinical pipeline is robust. Even in the absence of a clear clinical proof-of-principle, the prospects for ER-β agonist development seem promising.
4 Selective Androgen Receptor Modulators
Androgen Receptor is widely expressed in the body, and steroidal androgens, the important male circulating hormones, play important roles in maintaining bone strength, increasing muscle mass, and promoting the development of secondary sexual characteristics. However, virilizing side effects and limitations with regard to their route of administration have limited the clinical use of testosterone and closely related steroidal analogues and spurred interest in the development of SARMs. Testosterone preparations currently available in the US are administered either transdermally or intramuscularly. Transdermal preparations may cause skin sensitivity or transference of the androgen to individuals other than the patient (Wilson et al. 1998). Intramuscular testosterone often provides supraphysiologic levels of testosterone of unknown safety significance (Dhar et al. 2005). Orally available steroid androgens may pose a risk of hepatotoxicity (Boada et al. 1999).
In addition to the recognized risks of testosterone therapy including acne, edema, dyslipidemia, benign prostatic hypertrophy, and worsening of sleep apnea, the major concerns remain to be virilization in females, prostate cancer and cardiovascular toxicity in males (Osterberg et al. 2014; Vigen et al. 2013; Tan and Teoh 2013). The putative beneficial effects of testosterone therapy in female and certain male populations appear to be outweighed by the risks of virilization and unknown cardiovascular effects. The beneficial effects and success of commercially available SERMs have created interest to develop a therapeutic paradigm using tissue-specific SARMs that may act as full or partial agonists and antagonists for the AR in different tissues.
SARMs can be divided into four categories based on the structure of their pharmacologically active portions: aryl-propionamides, bicyclic hydantions, quinolines, and tetrahydroquinolones (Fig. 4). The pharmacokinetics and AR specificities of the compound in each of these categories differ widely (Gao et al. 2006). These two qualities, along with their tissue selectivity may give SARMs therapeutic advantages over steroidal androgens and create the possibility for SARMs targeting specific disease states or tissues.
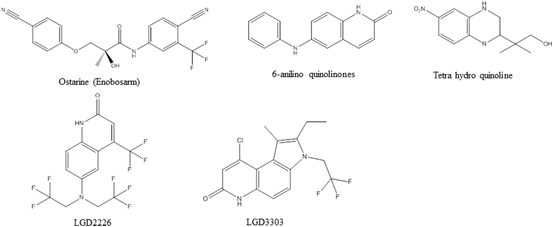
Fig. 4
Selective androgen receptor modulators (SARMs)
The tissue selectivity of a SARM in preclinical studies is typically evaluated in the Hershberger assay, where anabolic activity on muscle is compared to androgenic effects on prostate and seminal vesicles. While steroidal androgens such as DHT or testosterone non-selectively increase the size and weight of both muscle and prostate or seminal vesicles of castrated rats, SARMs only increase the muscle mass, while sparing prostate and seminal vesicles (Yin et al. 2003; Kim et al. 2005; Schlienger et al. 2009; Nique et al. 2012; Nagata et al. 2014). The same phenomenon is expected in humans too, where SARMs have been shown to increase lean body mass and physical function without affecting serum levels of prostate specific antigen (PSA) in men or causing hair-growth or other signs of virilization in women (Dalton et al. 2013; Dobs et al. 2013).
The field of SARMs is at an early stage, as none have been approved for clinical use. However, several companies have reported the results of preclinical and clinical studies with SARMs. The various pharmacophores, along with their demonstrated activity and potential clinical utility, are discussed below.
4.1 GTx Inc.
GTx, Inc. has published extensive results on the preclinical pharmacology and potential therapeutic applications of aryl propionamide SARMs. Enobosarm (Fig. 4; also referred to as S-22, GTx-024 and ostarine in the literature), their lead aryl propionamide SARM, is the most advanced clinical candidate. Enobosarm demonstrated exciting data in proof-of-concept Phase II clinical trials. A phase II double blind, randomized, placebo-controlled trial in elderly men and postmenopausal women and a phase II, double blind, randomized, placebo-controlled trial in men and women suffering from cancer cachexia have been reported (Dalton et al. 2013; Dobs et al. 2013). Without a prescribed diet or exercise regimen, all subjects treated with enobosarm had a dose-dependent increase in total lean body mass (LBM), with the 3 mg/day cohort achieving an increase of more than 1 kg compared to baseline and placebo after 3 months of treatment. Treatment with enobosarm also resulted in a dose-dependent improvement in functional performance measured by a stair climb test, with the 3 mg/day cohort achieving clinically significant improvement in speed and power. The SARM also demonstrated a favorable safety profile with no serious adverse events reported and no clinically significant changes in measurements for serum PSA (prostate), sebum production (skin and hair), or serum LH (pituitary) compared to placebo. Interestingly, subjects treated with 3 mg/day of enobosarm had a decline in fasting blood glucose, reduction in insulin levels, and reduction in insulin resistance (homeostasis model assessment) as compared to baseline, suggesting that SARMs might have therapeutic potential in diabetics or people at risk for diabetes. Phase I clinical studies with enobosarm showed that it was rapidly absorbed after oral administration with a half-life of about 1 day. Phase III trials to examine the safety and efficacy of enobosarm to prevent and treat muscle wasting in patients with stage III or IV non-small cell lung cancer were completed in 2013, and will be the subject of forthcoming reports by the investigators. Preliminary reports suggest that enobosarm was very well tolerated and demonstrated promising efficacy on LBM, but produced mixed results on stair climb power depending on the type of chemotherapy that the patients received.
Related posts:
 Multi-Functional Androgen Receptor Coregulator and Proto-Oncogene Melanoma Antigen-A11 (MAGE-A11)
Multi-Functional Androgen Receptor Coregulator and Proto-Oncogene Melanoma Antigen-A11 (MAGE-A11)
 Determines the Difference in DNA Binding Between the Androgen and the Glucocorticoid Receptors?
Determines the Difference in DNA Binding Between the Androgen and the Glucocorticoid Receptors?
 and Regulation of Nuclear Receptor Corepressor Complexes
and Regulation of Nuclear Receptor Corepressor Complexes
 Receptor Structure and Function
Receptor Structure and Function
 Analysis of Heterodimeric Nuclear Receptors
Analysis of Heterodimeric Nuclear Receptors
 Regulation and Intrinsic Disorder in Nuclear Hormone Receptors
Regulation and Intrinsic Disorder in Nuclear Hormone Receptors
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


