Bone-Forming (Osteogenic) Lesions
Disregarding, whether benign or malignant, the most characteristic feature of bone-forming neoplasms is the formation of osteoid tissue or mature bone directly by the tumor cells.
A. BENIGN BONE-FORMING LESIONS
Osteoma
Although not regarded as a neoplastic lesion by the World Health Organization, osteoma is included in this chapter because traditionally it has been regarded as a bone-forming lesion and because of its importance in differential diagnosis of osteoblastic tumors.
Definition:
Slow-growing osteoblastic surface lesion.
Epidemiology:
Fourth and fifth decades of life; men and women equally affected.
Sites of Involvement:
Occurs most commonly in skull and facial bones, including mandible, maxilla, frontal sinuses, ethmoid sinuses, paranasal sinuses, orbital bones, and calvarium; rarely involves the clavicles and long bones.
Clinical Findings:
On surface of bone—asymptomatic. If located in the paranasal sinuses, may cause mucocele, sinusitis, nasal discharge, and headache.
Orbital tumors may produce diplopia, exophthalmos, and blindness.
Presence of osteoma of long bones or multiple osteomas may be associated with Gardner syndrome (osteomas, colonic polyposis, skin fibromatoses, desmoids tumors, and epidermal and sebaceous cysts of skin).
Imaging:
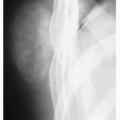
Radiography shows a homogenously dense, ivory-like sclerotic mass with sharply demarcated smooth borders, occasionally lobulated, attached to the cortex (Fig. 2.1A,B).
Computed tomography (CT) demonstrates a surface lesion without cortical invasion (Fig. 2.1C,D).
Magnetic resonance imaging (MRI) shows a lesion exhibiting low-signal intensity on all imaging sequences.
Pathology:
Gross (Macroscopy):
Nodular or dome-shaped mass, containing dense cortical bone (Fig. 2.2A).
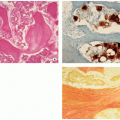
Histopathology:
Three types:
Cancellous (cancellous trabecular architecture with fatty marrow in the intertrabecular spaces; woven bone with plait of collagen fibers; roundish spindle-shaped osteocyte lacunae) (Fig. 2.2B,C).
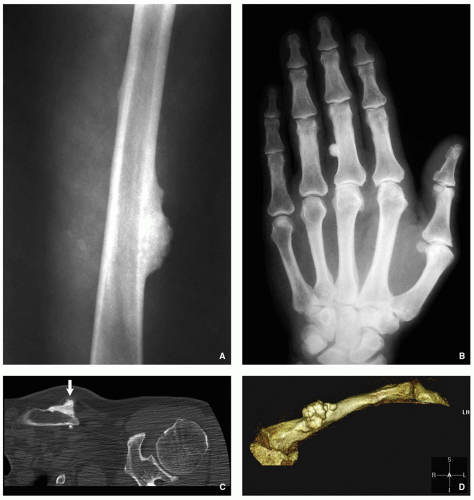
FIGURE 2.1 Osteoma. (A) Anteroposterior radiograph of the femur shows a sclerotic, ivory-like homogenous mass attached to the medial cortex. (B) Dorsovolar radiograph of the right hand shows a small homogenously sclerotic mass attached to the medial cortex of the proximal phalanx of the middle finger. (C) Coronal CT image of the left shoulder shows a sclerotic mass attached to the clavicle (arrow). Note lack of cortical invasion. (D) Three-dimensional CT image of the same patient as in (C) shows slightly lobulated mass on the surface of the clavicle.
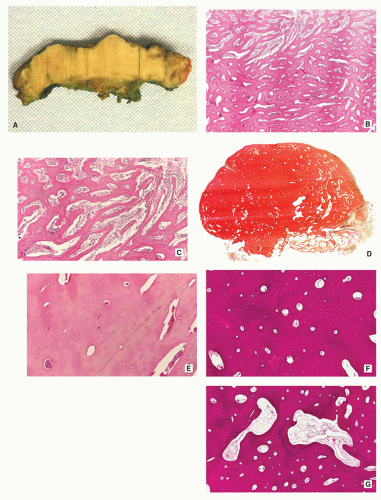
FIGURE 2.2 Osteoma. (A) Gross specimen of the osteoma of the clavicle. Lobulated mass is composed of a dense cortical bone. (B) Histopathology of cancellous variant. Irregularly formed bone trabeculae consist mostly of lamellar bone (H&E, original magnification ×25). (C) Cancellous variant. Intertrabecular spaces enclose highly vascular fibrous marrow tissue (H&E, original magnification ×20). (D) Compact variant. Dense, compact, mostly lamellar bone surrounds narrow marrow spaces (van Gieson, whole-mount section). (E) Compact variant. Some osteocyte lacunae are empty as a result of osteocyte decline due to poor nourishment (H&E, original magnification ×200). (F,G) Mixed variant. Note areas of compact and cancellous architecture (H&E, original magnification ×200).
Compact (dense, compact, mature lamellar bone; no Haversian systems) (Fig. 2.2D,E).
Mixed (admixed areas of compact and cancellous architecture) (Fig. 2.2F,G).
Prognosis:
No recurrence after surgical excision.
Differential Diagnosis:
▪ Parosteal osteosarcoma
Irregular outer contour.
Commonly lack of homogeneity, periphery of the tumor may be less dense than the center.
Occasionally invasion of the cortex by tumor.
Incomplete cleft between the tumor and adjacent cortex commonly present.
Histopathology shows streamers of woven to woven-lamellar bone with heavy collagenized stroma. Moderately cellular foci with nuclei exhibiting slight pleomorphism.
▪ Sessile osteochondroma
Cortex of the host bone merges without interruption with cortex of the lesion, and respective cancellous portions of adjacent bone and osteochondroma communicate.
Histopathology shows cartilaginous cap composed of hyaline cartilage arranged similarly to growth plate. Beneath it, zone of endochondral ossification with vascular invasion and replacement of calcified cartilage by newly formed bone.
▪ Well-matured focus of parosteal myositis ossificans
Zonal phenomenon (periphery of the lesion more mature than the center).
Radiolucent cleft separates the lesion from adjacent cortex.
Regression of the lesion with time.
Histopathology shows trabecular bone and fibrous marrow. Histologic zonal phenomenon consists of immature bone in the center with proliferating osteoblasts, fibroblasts, and areas of hemorrhage and necrosis; mature bone on periphery.
▪ Monostotic form of melorheostosis (forme fruste)
Wavy outline of cortical thickening resembling wax dripping down one side of a candle.
Occasionally, lesion extends into the medullary portion of bone.
Histopathology shows thickened cortical bone containing irregularly arranged Haversian canals surrounded by cellular fibrous tissue. Osteoblastic activity usually present.
Osteoid Osteoma
Osteoid osteoma is a benign osteoblastic lesion characterized by a nidus of osteoid tissue, which may be purely radiolucent or have a sclerotic center. The nidus has a limited growth potential and usually measures less than 2 cm in diameter. It is often surrounded by a zone of reactive bone formation. Very rarely, an osteoid osteoma may have more than one nidus, in which instant it is called a multicentric or multifocal osteoid osteoma. Depending upon its location in a particular part of the bone, the lesion is classified as cortical, medullary (cancellous), or subperiosteal. Osteoid osteoma can be further subclassified as extracapsular or intracapsular (intra-articular).
Definition:
Benign bone-forming tumor.
Similar to osteoblastoma but smaller in size (nidus 1.5 to 2.0 cm).
Epidemiology:
Usually occurs in second or third decade.
Male-to-female ratio of 3:1.
Sites of Involvement:
Most common in long bones—femur/tibia (cortex of metaphysis).
May be found in any bone.
Rare cases in ethmoid bone have been reported.
Clinical Findings:
Intense localized pain particularly at night.
Pain relieved by aspirin, nonsteroidal anti-inflammatory drugs (NSAIDs), surgery, or radiofrequency ablation.
When osteoid osteoma is present in the small bones of the hands and feet, patients are typically misdiagnosed and treated for an inflammatory process (such as osteomyelitis, arthritis).
Imaging:
Radiography:
Small, round radiolucency, surrounded by zone of sclerosis (particularly in cortical location) (Fig. 2.3A,B).
Small, round radiolucency without zone of sclerosis (particularly in cancellous bone) (Fig. 2.3C).
Small sclerotic or radiolucent lesion without zone of sclerosis, but with minimal periosteal response (in subperiosteal location) (Fig. 2.3D).
If lesion localized within the joint (intracapsular)— periarticular osteoporosis and precocious osteoarthritis (Fig. 2.3E).
Periosteal reaction (in intracortical location) (see Fig. 2.3B).
Scintigraphy:
Increased activity of the radiopharmaceutical agent on both immediate and delayed images (Fig. 2.5).
Characteristic “double-density” sign (Fig. 2.6).
Computed Tomography:
Better characterization of the lesion (Fig. 2.7).
Measurements of the nidus can be obtained.
Vascular groove sign.
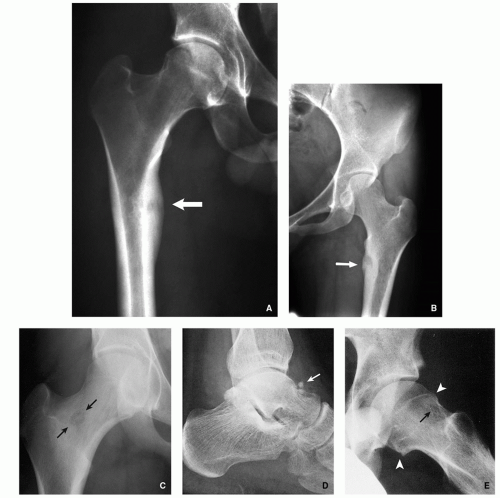 FIGURE 2.3 Radiography of osteoid osteoma. (A) Anteroposterior radiograph of the right hip of a 27-year-old man shows a radiolucent lesion within the medial femoral cortex associated with peripheral sclerosis (arrow). (B) Anteroposterior radiograph of the left hip of a 22-year-old woman shows similar radiolucent nidus surrounded by sclerotic reaction in the medial femoral cortex (arrow). (C) Intramedullary nidus within the femoral neck (arrow) shows no evidence of reactive sclerosis. (D) Subperiosteal lesion on the surface of the talus (arrow) show minimal periosteal reaction and no evidence of reactive sclerosis. (E) Intracapsular lesion is present in the left femoral neck (arrow) without evident perilesional sclerosis in a 14-year-old boy. Observe periarticular osteoporosis and early osteoarthritic changes in form of osteophytes (arrowheads). |
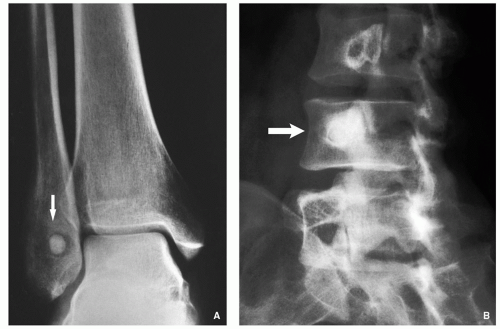 FIGURE 2.4 Radiography of osteoid osteoma. (A) Anteroposterior radiograph of the ankle shows a radiolucent lesion within the distal fibula with sclerotic center (arrow). (B) Oblique radiograph of the lumbar spine shows purely sclerotic nidus of osteoid osteoma affecting the pedicle of L4 (arrow). |
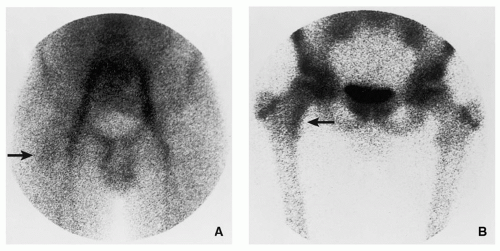 FIGURE 2.5 Scintigraphy of osteoid osteoma. (A) In the first phase of a three-phase radionuclide bone scan, 1 minute after intravenous injection of 15 mCi (555 MBq) technetium-99m-labeled MDP, there is increased activity in the iliac and femoral vessels. Discrete activity in the area of the medial left femoral neck (arrow) is related to the nidus of osteoid osteoma. (B) In the third phase, 2 hours after injection, there is accumulation of a bone-seeking tracer in the femoral neck lesion (arrow). (Reprinted from Greenspan A. Benign bone-forming lesions: osteoma, osteoid osteoma, and osteoblastoma. Skeletal Radiol. 1993;22:490-500, with permission.) |
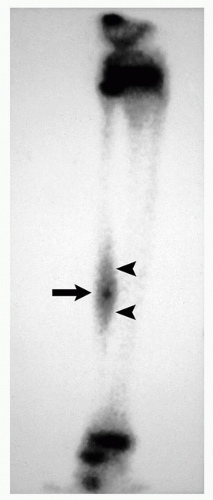 FIGURE 2.6 Scintigraphy of osteoid osteoma. Radionuclide bone scan shows a classic “double density” sign of osteoid osteoma located in the tibia: markedly increased radioactivity in the center (arrow) is related to the nidus, less active areas (arrowheads) represent reactive sclerosis. |
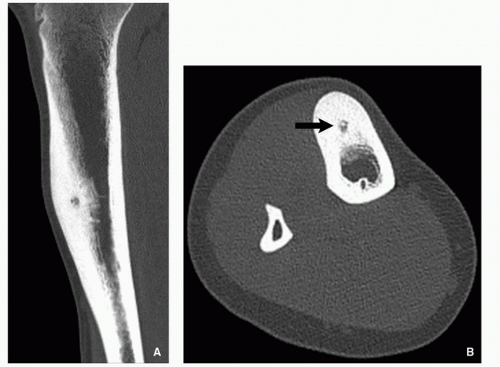 FIGURE 2.7 Computed tomography of osteoid osteoma. (A) Coronal reformatted CT image of the tibia and (B) axial CT of the same patient show a well-defined low-attenuation nidus with sclerotic center located in the anterior cortex (arrow). |
Ultrasound:
Focal cortical irregularity and adjacent hypoechoic synovitis at the site of intra-articular lesion.
Magnetic Resonance Imaging:
High-signal intensity of the nidus on T2-weighted and inversion recovery (IR) sequences (Fig. 2.8).
Occasionally confusing pattern, may be mistaken for inflammation or malignancy.
Pathology:
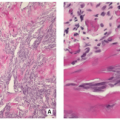
Gross (Macroscopy):
Small, cortically based, red, gritty round lesion (Fig. 2.9).
Histopathology:
Limited growth pattern (1.5 to 2.0 cm).
Sharp circumscription of the nidus near cortical surface (Fig. 2.10A).
Nidus composed of anastomosing bone trabeculae with variable mineralization (Fig. 2.10B).
Bone trabeculae lined by plump osteoblasts (Fig. 2.10C).
Osteoblastic and osteoclastic activities often prominent.
Vascularized connective tissue, surrounded by sclerotic bone displaying a variety of maturation patterns.
Benign giant cells may be present.
Genetics:
Described loss of 17q.
Structural alterations of chromosome 22 – [del(22)(q13.1)].
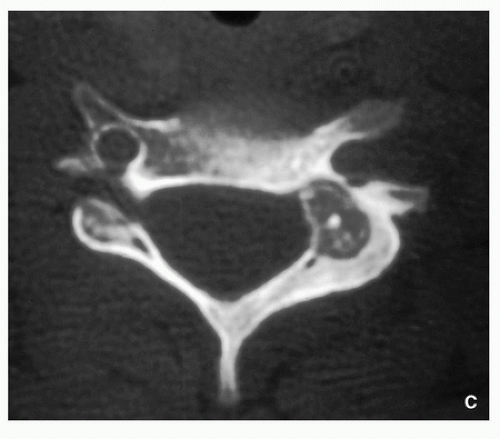 FIGURE 2.7 (Continued). (C) Axial CT image of the cervical vertebra C6 of another patient shows a low-attenuation nidus with a sclerotic center, located in the left pedicle and extending to the lamina. (C—Reprinted with permission from Greenspan A. Benign bone-forming lesions: osteoma, osteoid osteoma, and osteoblastoma. Skeletal Radiol. 1993;22:490-500). |
 FIGURE 2.8 Magnetic resonance imaging of osteoid osteoma. (A) Sagittal and (B) axial T2-weighted MR images show a highsignal-intensity nidus in posterolateral cortex of tibia (arrows). |
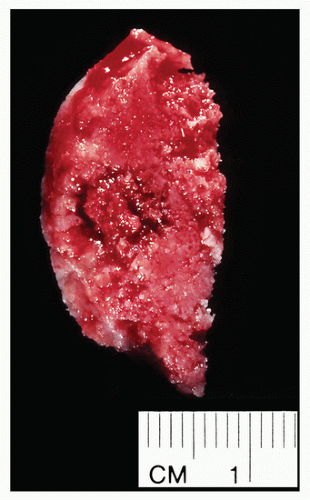 FIGURE 2.9 Osteoid osteoma, gross specimen. Note wellcircumscribed nidus exhibiting hypervascular zone with surrounding sclerotic rim. |
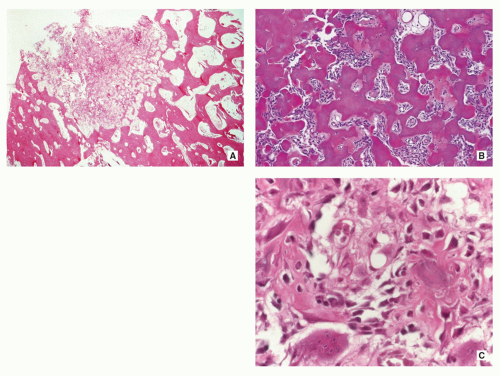 FIGURE 2.10 Osteoid osteoma, histopathology. (A) Lowpower magnification shows a well-demarcated nidus composed of trabeculae and immature woven bone (center), surrounded by sclerotic bone (H&E, original magnification ×6). (B) Higher magnification of the center of the nidus shows interconnected, ossified bone trabeculae within areas of vascularized connective tissue, and osteoblastic activity (H&E, original magnification ×100). (C) High-power photomicrograph of the nidus shows bone trabeculae rimmed by osteoblasts (H&E, original magnification ×400). |
Complications:
Accelerated bone growth if nidus located near the growth plate.
Painful scoliosis if nidus located within the vertebra (particularly within the neural arch).
Precocious arthritis if lesion located within the joint (intracapsular).
Recurrence if lesion not completely excised.
Prognosis:
Excellent, after total surgical excision or radiofrequency thermal ablation (RFTA).
Differential Diagnosis:
▪ Osteoblastoma
Larger nidus (more than 2 cm). Less reactive sclerosis, but more prominent periosteal reaction.
Histopathology is similar to osteoid osteoma, but less organized pattern of osteoid and reticular bone distribution; occasionally, spindle-shaped hyperchromatic cells with uniform nuclei and irregular eosinophilic cytoplasm interdispersed between immature bone trabeculae.
▪ Bone abscess
Serpentine radiolucent track extending from the lesion.
May cross the growth plate.
Histopathology shows inflammatory cells and areas of necrosis.
▪ Bone island (enostosis)
“Thorny radiations” (pseudopodia) at periphery of the lesion (brush borders) that blend with trabeculae of the host bone.
Usually no significant activity on scintigraphy.
Histopathology shows focus of mature compact bone; wide bands of parallel or concentric lamellae; marrow spaces resemble haversian canals.
▪ Stress fracture
More linear radiolucency with perpendicular or oblique orientation.
Osteoblastoma
Osteoblastoma is a benign bone-forming tumor, similar to osteoid osteoma, but larger in size. Natural history differs
from that of osteoid osteoma: whereas the latter lesion tends toward regression, osteoblastoma tends toward progression, and even malignant transformation, although this possibility remains controversial. Toxic osteoblastoma, a rare variant of this tumor, has also been reported. It is associated with systemic manifestations, including diffuse periostitis of multiple bones, fever, and weight loss. Although the long bones are commonly affected by osteoblastoma, the lesion has a predilection for the vertebral column.
from that of osteoid osteoma: whereas the latter lesion tends toward regression, osteoblastoma tends toward progression, and even malignant transformation, although this possibility remains controversial. Toxic osteoblastoma, a rare variant of this tumor, has also been reported. It is associated with systemic manifestations, including diffuse periostitis of multiple bones, fever, and weight loss. Although the long bones are commonly affected by osteoblastoma, the lesion has a predilection for the vertebral column.
Definition:
Benign bone-forming neoplasm producing woven bone spicules bordered by prominent osteoblasts (measuring more than 2.0 cm).
Epidemiology:
About 1% of all primary bone tumors, and 3% of all benign bone tumors.
Age: 10 to 30 years (but predominantly teenagers).
Male-to-female ratio of 2.5:1.
Sites of Involvement:
Predilection for the spine (40% to 55% of cases).
Other common sites—femur and proximal tibia.
Cementoblastoma of the jaw is considered osteoblastoma and is attached to the root of tooth.
Clinical Findings:
Dull, localized pain, rarely interfered with sleep.
Tenderness on palpation at the tumor site.
Lesion in the spine may cause back pain, scoliosis, and nerve root compression.
Jaw lesion may produce tooth pain.
Aspirin does not relieve pain.
Tendency to progression (question of malignant transformation, a controversial issue).
Imaging:
Distinctive four types of the lesion:
Giant osteoid osteoma (Fig. 2.11A-C).
Lytic expansive lesion, similar to aneurysmal bone cyst (ABC), with central mineralization (usually spine lesions) (Fig. 2.11D,E).
Aggressive lesion simulating a malignant tumor (Fig. 2.11F-H).
Juxtacortical (periosteal) in location (Fig. 2.11I-L).
General imaging features:
Radiolucent well-circumscribed oval or round lesion.
May or may not exhibit perilesional sclerosis.
Prominent periosteal reaction.
Scintigraphy invariably demonstrates intense focal accumulation of radiopharmaceutical.
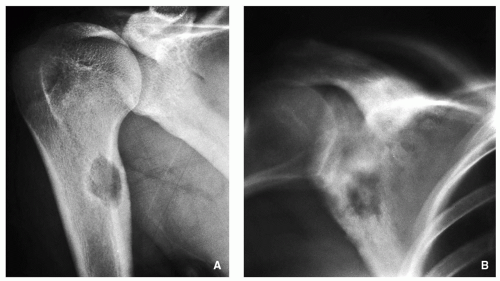
FIGURE 2.11 Radiography of osteoblastoma. “Giant” osteoid osteoma-like lesions. Note that all three lesions are very similar to osteoid osteoma, but much larger, and periosteal reaction is more prominent (A-C). Aneurysmal-like expansion with central radiodensities. This type of presentation is usually seen in the spine lesions.
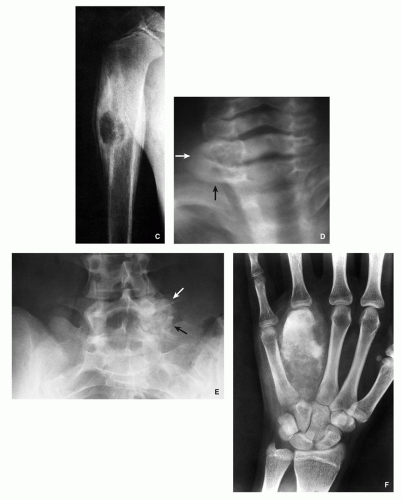
FIGURE 2.11 (Continued). (D) Expansive blow-out lesion with central opacities is seen in the lamina of C6 (arrows) and (E) in the lamina, pedicle, and transverse process of L5 (arrows). Aggressive lesion mimicking malignant tumors. (F) The lesion affecting the forth metacarpal bone completely destroyed the bone, having an appearance of osteosarcoma.
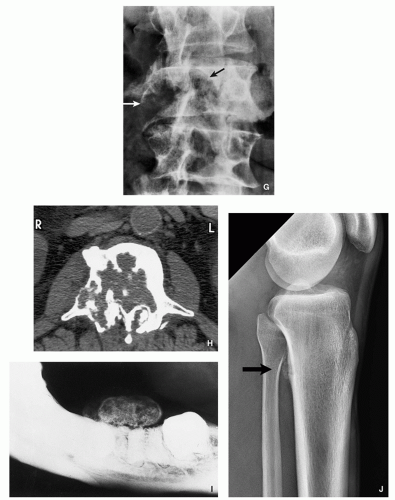
FIGURE 2.11 (Continued). (G,H) Aggressive osteoblastoma destroyed part of the vertebral body of L3 (arrows). Periosteal location. (I) Juxtacortical lesion of the mandible.
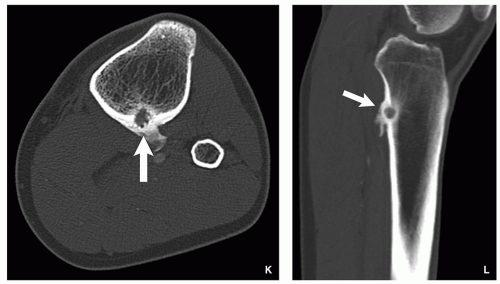
FIGURE 2.11 (Continued). (J-L) Juxtacortical location in the posterior cortex of proximal tibia, invading the endocortex and evoking periosteal reaction (arrows). (D,E,G,H—Reprinted with permission from Greenspan A. Benign bone-forming lesions: osteoma, osteoid osteoma, and osteoblastoma. Skeletal Radiol. 1993;22:494-500).
CT better characterizes the lesion and allows to obtain measurements (Fig. 2.12).
MRI shows extensive peritumoral bone marrow edema, and low signal intensity on all sequences when lesion is heavily mineralized (exhibiting predominantly osteoblastic matrix) (Fig. 2.13), but high signal intensity on T2-weighted and IR sequences in less mineralized (radiolucent) lesions.
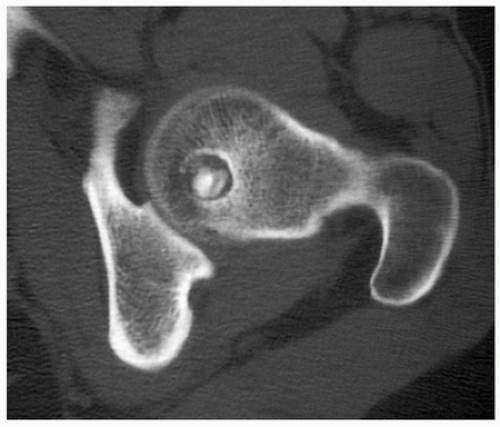 FIGURE 2.12 Computed tomography of osteoblastoma. Axial CT image of the left hip of a 20-year-old man shows a lowattenuation lesion with sclerotic center within the femoral head, measuring 2.75 cm. |
Pathology:
Gross (Macroscopy):
Central nidus is red, soft, and friable (vascular).
Often with gritty or sandpaper consistency, if calcified may be yellow and gritty.
Cortical bone may be thin-out or destroyed, occasionally with cyst formation representing formation of secondary ABC.
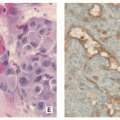
Histopathology:
Nidus appears well circumscribed, with tumor osteoid merging with adjacent uninvolved bone.
Composed of woven bone spicules or trabeculae lined by a single layer of osteoblasts (Fig. 2.14A).
Irregular interlacing network of osteoid with prominent osteoblastic rimming and features of woven bone (Fig. 2.14B,D).
Osteoblasts exhibit benign cytologic features, although increased mitotic activity without atypical forms may be present.
Osteoid may be fine and lace-like with variable mineralization, separated by fibrovascular stroma containing multinucleated osteoclast-like giant cells (Fig. 2.14C).
Rich vascularity of stroma (see Fig. 2.14A).
Large blood lakes representing secondary aneurysmal cystic changes may be seen (Fig. 2.14E).
Scattered osteoclast-type multinucleated giant cells are often present (see Fig. 2.14C).
Focal blood-filled spaces mimicking ABC may be present (Fig. 2.14E).
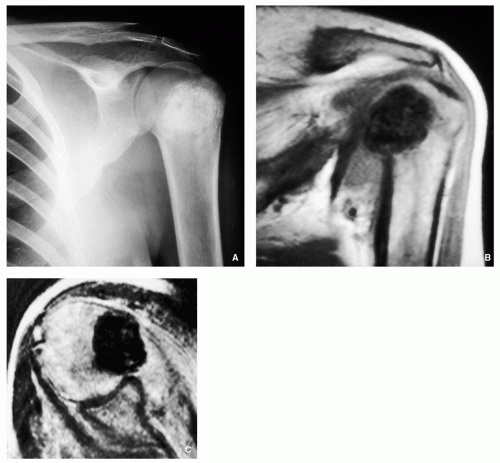
FIGURE 2.13 Magnetic resonance imaging of osteoblastoma. (A) Anteroposterior radiograph of the left shoulder shows a large sclerotic lesion within the humeral head. (B) Coronal T1-weighted MR image shows the lesion to be of low-signal intensity. (C) Axial spin-echo T2-weighted MRI shows that the lesion remains of low-signal intensity, indicating osteoblastic matrix.
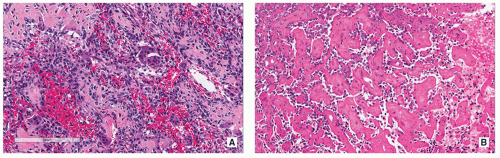
FIGURE 2.14 Histopathology of osteoblastoma. (A) Low-power photomicrograph shows irregular trabeculae of woven bone (osteoid) surrounded by densely arranged osteoblasts and some giant cells. Note also several ectatic blood vessels (H&E, original magnification ×50). (B) At higher magnification, the osteoblastic rimming of the trabeculae is conspicuous (H&E, original magnification ×100).
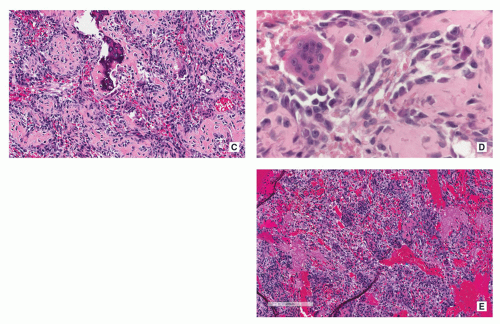
FIGURE 2.14 (Continued). (C) Densely arranged small trabeculae made up of woven bone are surrounded by osteoblasts. In the narrow marrow spaces, some capillaries and giant cells of the osteoclast type are seen (H&E, original magnification ×200). (D) High-power photomicrograph reveals osteoblasts and osteoclasts on the surface of the immature bone trabeculae (H&E, original magnification ×400). (E) Trabeculae of woven bone rimmed with osteoblasts and large blood lakes representing secondary aneurysmal cystic changes are present (H&E, original magnification ×100).
Osteoblastomas do not infiltrate and isolate preexisting lamellar bone structures as does osteosarcoma.
Cartilage usually not present.
Epithelioid features may be seen represented by large cells with abundant eosinophilic cytoplasm and enlarged nuclei containing large nucleoli.
Rare cytologically atypical multinucleated giant cells without mitotic activity may be seen (tumors with these features may be designated bizarre osteoblastoma or pseudomalignant osteoblastoma).
Prognosis:
Excellent.
Recurrences unusual if totally excised.
Differential Diagnosis:
▪ Osteoid osteoma
Smaller nidus (less than 2 cm).
More reactive sclerosis.
Vascular groove sign on CT.
Different clinical presentation (pain relief by salicylates).
Histopathology similar to osteoblastoma, but at the periphery of the nidus, a fibrovascular rim is present, and nidus itself exhibits a distinct zonal pattern with central maturation to more mineralized woven bone.
▪ Bone abscess (Brodie abscess)
Serpentine radiolucent track extending from the lesion.
May cross the growth plate.
Histopathology shows inflammatory cells and areas of necrosis.
▪ Aneurysmal bone cyst
More prominent expansion and ballooning.
Buttress of periosteal reaction.
Histopathology shows multiple blood-filled sinusoid spaces separated by fibrous septae displaying lamellae of primitive woven bone; may contain hemosiderin and reactive foam cells; solid areas composed of fibrous elements contain irregular bone trabeculae and giant cells.
▪ Enchondroma (in short tubular bone)
Central chondroid calcifications.
Lack of periosteal reaction (unless pathologic fracture).
Histopathology shows lobules of hyaline cartilage of variable cellularity with evidence of endochondral ossification at the periphery of the lobules; the tissue is sparsely cellular, and the cells containing small dark-staining nuclei are located in the lacunae; intercellular matrix has a uniform translucent appearance and contains relative little collagen; matrix calcifications may be present.
▪ Osteosarcoma
More aggressive presentation.
Interrupted periosteal reaction (sunburst, lamellated, Codman triangle).
Make look very similar to aggressive osteoblastoma.
Histopathology shows permeation of cortical bone; attenuation and “trapping” of lamellar bone; atypical mitoses and anaplasia of cells; hyperchromatism and pleomorphism of cells and nuclei; tumor bone and tumor cartilage formed by malignant cells.
B. MALIGNANT BONE-FORMING TUMORS
Osteosarcomas
Osteosarcomas, the most common primary malignant tumors of bone, comprise a family of connective tissue tumors with various degrees of malignant potential. There are several types of osteosarcoma (Fig. 2.15), each having distinctive clinical, imaging, and histopathologic characteristics. The common feature of all types is that the osteoid and bone matrix are formed directly by the malignant cells of connective tissue. The majority of osteosarcomas are of unknown cause and therefore are referred to as primary or idiopathic. A smaller number of tumors are related to known factors predisposing to malignancy, such as Paget disease, fibrous dysplasia, external ionizing irradiation, or ingestion of radioactive substances. These lesions are referred as to as secondary osteosarcomas. All types of osteosarcomas may further be subdivided by anatomic sites into tumors of appendicular skeleton and axial skeleton, as well as may be classified on the basis of their location in the bone as central (intramedullary), intracortical, and juxtacortical. A separate group consists of primary osteosarcomas originating in soft tissues (extraskeletal or soft-tissue osteosarcomas). Finally, yet another group comprises metastatic lesions (in the lungs, bones, and soft tissues).
It is important to mention here that there are numerous genetic disorders, marked by chromosome instability, associated with the development of various tumors, including osteosarcomas. Among these rare conditions are Rothmund-Thomson syndrome, Werner syndrome, Li-Fraumeni syndrome, retinoblastoma syndrome, and Bloom syndrome.
Histopathologically, osteosarcomas are graded on the basis of their cellularity, nuclear pleomorphism, and degree of mitotic activity (Table 2.1). The grading has clinical, therapeutic, and prognostic values.
Conventional Osteosarcoma
Definition:
Primary intramedullary high-grade malignant sarcoma in which neoplastic cells produce osteoid or bone.
Epidemiology:
Most common primary nonhematopoietic malignancy of bone.
Incidence in the United States is 4 to 5 per million individuals with 1,000 to 1,500 new cases diagnosed annually, which accounts for approximately 20% of all primary malignant tumors.
Most common in second decade of life with 60% of tumors in patients younger than 25 years.
About 30% occur in patients over 40 years of age (predisposing conditions include radiation therapy and Paget disease of bone).
Male-to-female ratio of 3:2.
Osteosarcoma in children 5 years and younger is very uncommon, accounting for less than 2% of osteosarcomas in the pediatric population.
Sites of Involvement:
Osteosarcoma most commonly involves long bones of the appendicular skeleton with preference to the distal femur, proximal tibia, and proximal humerus.
Most of the tumors are centered in metaphysis of long bones (90%), followed by diaphysis (9%), and rarely epiphysis.
Involvement of other sites than long bones (i.e., jaws, pelvis, spine, and skull) tends to increase with age.
Clinical Presentation:
Typically presents as progressively enlarging mass.
Pain is deeply seated and boring in nature, commonly noted months prior to the diagnosis, and usually increases in intensity over time.
Skin overlying the tumor may be warm, erythematous, edematous, with prominent engorged veins.
Large tumors near the joints may restrict the range of motion and produce joint effusion.
Advanced cases result in weight loss and cachexia.
Pathologic fracture through the destructive mass may be seen.
Imaging:
Radiography:
Variable radiographic appearances, reflecting histopathology; conventional tumors usually present as a large, destructive, poorly defined, mixed lytic and blastic lesions exhibiting wide zone of transition and moth-eaten bone destruction, accompanied by cortical invasion and extension into the soft tissues (Figs. 2.16 and 2.17). Purely sclerotic (osteoblastic) (Fig. 2.18A,B) and purely lytic (chondroblastic or fibroblastic) (Fig. 2.18C) lesions may also be encountered.
Tumor/periosteal interaction leads to variety of manifestations secondary to periosteal reaction (periosteal reactive bone formation), such as fine triangular structures (Codman triangle) (Fig. 2.19A), fine, ill-defined perpendicular spiculations (velvet type) (see Fig. 2.16A,B), perpendicular or radiating coarse striations (“hair-on-end”
or “sunburst” periosteal reaction) (Fig. 2.19B, see also Fig. 2.16A,B), or less common lamellated periosteal layers (“onion skin” type of periosteal reaction) (Fig. 2.19C).
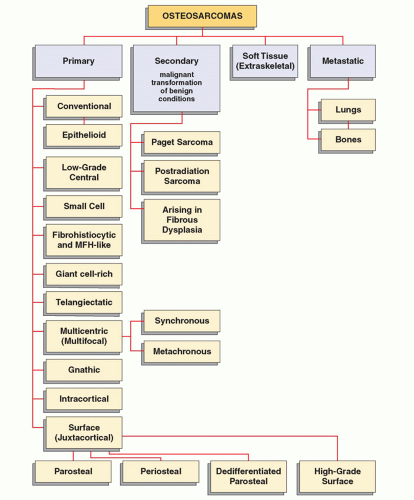 FIGURE 2.15 Classification of osteosarcomas. |
Scintigraphy:
Invariably increased uptake of radiopharmaceutical agent (Figs. 2.20 and 2.21); may show “skipped” lesions (Fig. 2.22).
Computed Tomography:
Demonstrates intramedullary and extracortical extension of the tumor and periosteal reaction (Fig. 2.23).
Accurately demonstrates cortical destruction.
Using Hounsfield values, one can distinguish between tumor tissue, peritumoral edema, and normal bone marrow.
TABLE 2.1 Histologic Grading of Osteosarcomaa | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||
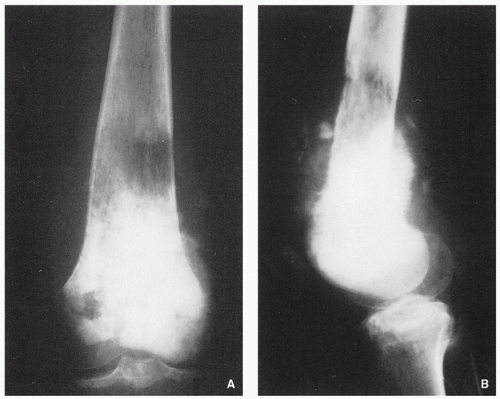 FIGURE 2.16 Radiography of conventional osteosarcoma. Anteroposterior (A) and lateral (B) radiographs shows the typical features of this tumor in the distal femur of a 19-year-old woman. Medullary and cortical destruction is present, associated with aggressive periosteal reaction of the velvet and sunburst types. A soft-tissue mass is present containing foci of tumor bone. |
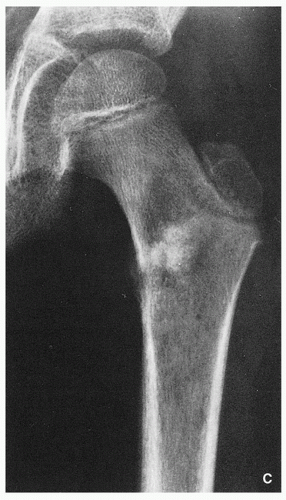 FIGRE 2.16 (Continued). (C) In another patient, a 12-year-old boy, anteroposterior radiograph of the left femur demonstrates a mixed, lytic, and sclerotic tumor in the intertrochanteric region. |
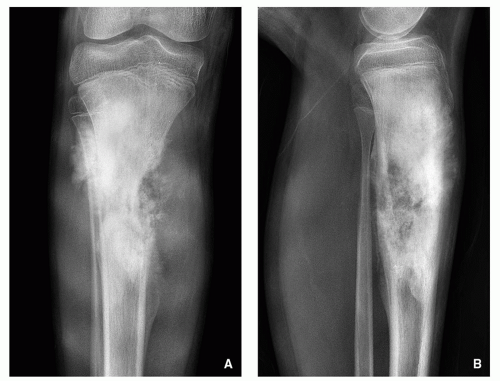 FIGURE 2.17 Radiography of conventional osteosarcoma. (A) Anteroposterior and (B) lateral radiographs of the right proximal leg of a 12-year-old boy show a sclerotic lesion affecting diaphysis of the tibia, associated with destruction of the medial cortex, aggressive periosteal reaction, and a soft-tissue mass. |
Magnetic Resonance Imaging:
Similarly to CT shows intramedullary extension of the tumor and well characterizes soft-tissue mass (Figs. 2.24, 2.25, 2.26 and 2.27).
Usually heterogenous low signal intensity on T1-weighted and heterogenous high signal on T2-weighted and other water-sensitive sequences (Fig. 2.28).
Allows distinction between the tumor and peritumoral edema (postcontrast studies using gadolinium).
Allows evaluation of involvement of neurovascular structures by the tumor.
Pathology:
Gross (Macroscopy):
Heterogeneous large fleshy mass with ossified and nonossified components and occasionally foci of cartilage (Fig. 2.29).
Commonly the tumor breaks through the cortex and is invading the adjacent soft tissues, where foci of tumor bone formation and hemorrhage may be seen (Fig. 2.30).
The tumor may invade the nearby joint (Fig. 2.31).
New bone formation (periosteal reaction) at periphery of the tumor.
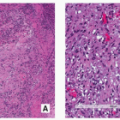
Histopathology:
Tumor cells show prominent atypia and pleomorphism and may exhibit epithelioid, plasmacytoid, fusiform, ovoid, small round, or spindled appearances (Fig. 2.32).
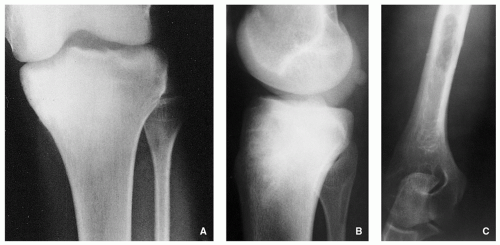
FIGURE 2.18 Radiography of conventional osteosarcoma. Anteroposterior (A) and lateral (B) radiographs demonstrate sclerotic variant of this tumor affecting proximal tibia. (C) Anteroposterior radiograph shows a destructive purely lytic lesion in the distal humerus, proven on histopathologic examination to represent fibroblastic osteosarcoma.
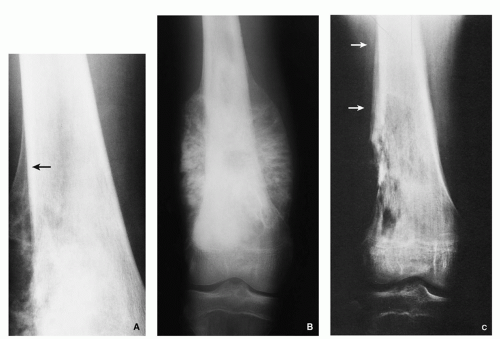
FIGURE 2.19 Radiography of periosteal reaction in osteosarcoma. (A) Typical Codman triangle (arrow). (B) Typical sunburst pattern. (C) Typical lamellated (onion skin) pattern (arrows).
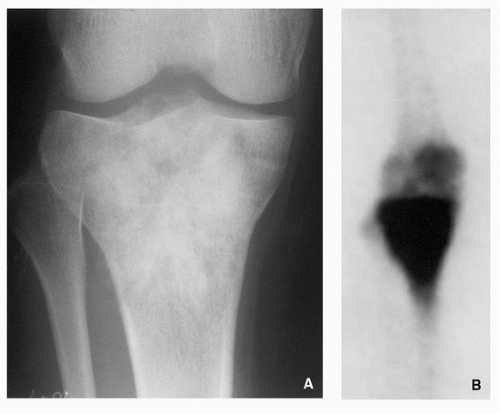
FIGURE 2.20 Scintigraphy of conventional osteosarcoma. (A) Anteroposterior radiograph of the right knee shows a sclerotic variant in the proximal tibia. (B) After intravenous injection of 15 mCi (555 MBq) of technetium-99m-labeled methylene diphosphonate, there is significant uptake of the radiopharmaceutical agent by the tumor.

FIGURE 2.21 Scintigraphy of conventional osteosarcoma. (A) Anteroposterior and (B) lateral radiographs of the right knee of a 13-year-old girl show a sclerotic lesion affecting the metaphysis and diaphysis of the tibia. (C) A total body radionuclide bone scan and
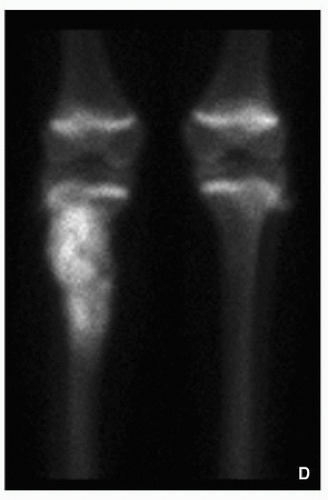
FIGURE 2.21 (Continued). (D) coned-down scintigraphic images of the knees show significantly increased uptake of the radiopharmaceutical agent by a tumor in the proximal right tibia.
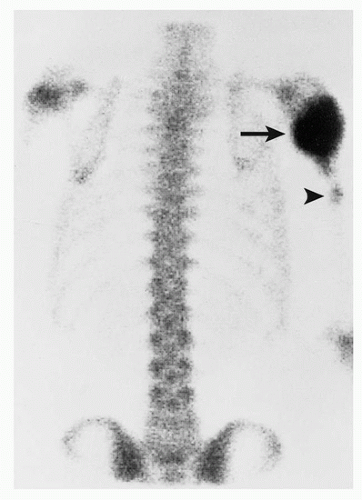
FIGURE 2.22 Scintigraphy of conventional osteosarcoma. In a 7-year-old-boy with a lesion in the proximal left humerus, radionuclide bone scan shows increased uptake of the tracer by the tumor (arrow). In addition, a small focus of activity (arrowhead) represents a skip lesion.
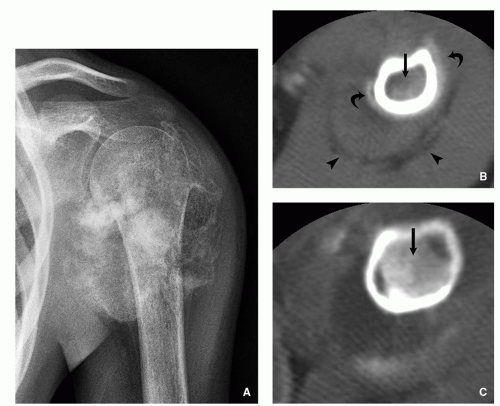
FIGURE 2.23 Computed tomography of conventional osteosarcoma. (A) Anteroposterior radiograph of the left shoulder shows a bone-forming tumor affecting proximal humerus associated with aggressive periosteal reaction and soft-tissue mass. (B,C) Two CT axial sections show high-attenuation tumor replacing the bone marrow (arrows), periosteal reaction (curved arrows), and soft-tissue mass (arrow heads).
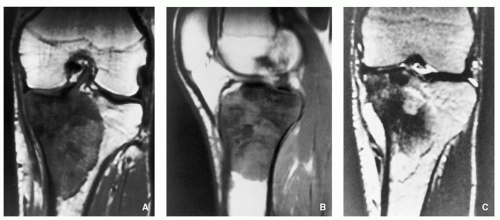
FIGURE 2.24 Magnetic resonance imaging of conventional osteosarcoma. (A) Coronal and (B) sagittal T1-weighted MR images of the knee of a 17-year-old boy show a heterogenous but predominantly low signal intensity appearance of the tumor affecting proximal tibia. (C) On T2-weighted MR image, the sclerotic parts of the tumor remain of low signal intensity, whereas the more distal, nonmineralized tissue and soft-tissue mass exhibit high signal intensity.

FIGURE 2.25 Magnetic resonance imaging of conventional osteosarcoma. (A) Sagittal T2-weighted MR image of the knee shows a tumor affecting proximal tibia, exhibiting heterogenous mixed high and low signal intensities. Soft tissue extension of the tumor is not well depicted. Coronal (B) and axial (C) T1-weighted MR images obtained after intravenous injection of gadolinium show lack of enhancement of the sclerotic part of the tumor, but significant enhancement of the lytic part proximally. Note also enhancement of a soft-tissue mass containing tumor-bone imaged as low signal intensity foci.
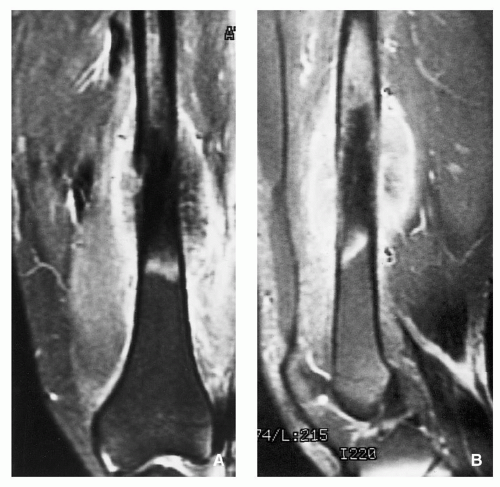
FIGURE 2.26 Magnetic resonance imaging of conventional osteosarcoma. Coronal (A) and sagittal (B) T1-weighted fat-suppressed MR images of a 29-yearold man obtained after intravenous injection of gadolinium show tumor enhancement in the medullary portion of the femur and in the soft-tissue mass. Heavy mineralized portion of the tumor does not enhance and remains of low signal intensity.
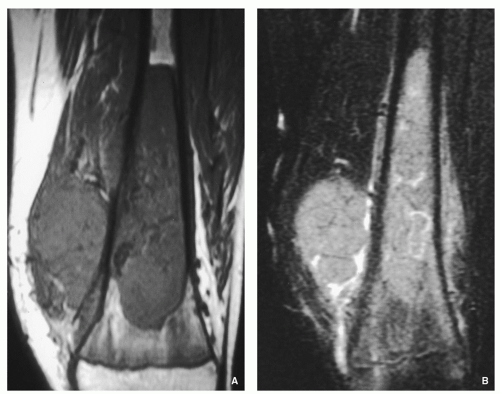
FIGURE 2.27 Magnetic resonance imaging of chondroblastic osteosarcoma. (A) Coronal T1-weighted MR image of the distal femur of a 14-year-old boy with chondroblastic osteosarcoma shows a large low-signal-intensity tumor in the bone marrow associated with a soft-tissue mass. (B) After intravenous administration of gadolinium, there is diffuse enhancement of both intramedullary tumor and soft-tissue mass.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree

 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access