Chapter 3 Challenge
Albayram et al. 2002 Albayram S, Melham E, Mori S, et al. Holoprosencephaly in children: diffusion tensor MR imaging of white matter tracts of the brainstem. Initial experience. Radiology. 2002;223:645-651.
Barkovich et al. 2002 Barkovich A, Simon E, Clegg N, Kinsman S, Hahn J. Analysis of the cerebral cortex in holoprosencephaly with attention to the sylvian fissures. AJNR Am J Neuroradiol. 2002;23:143-150.
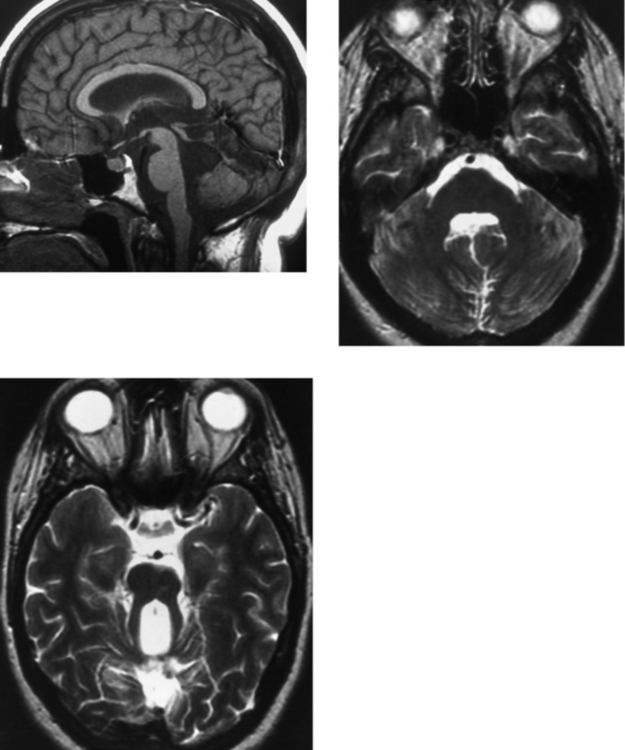
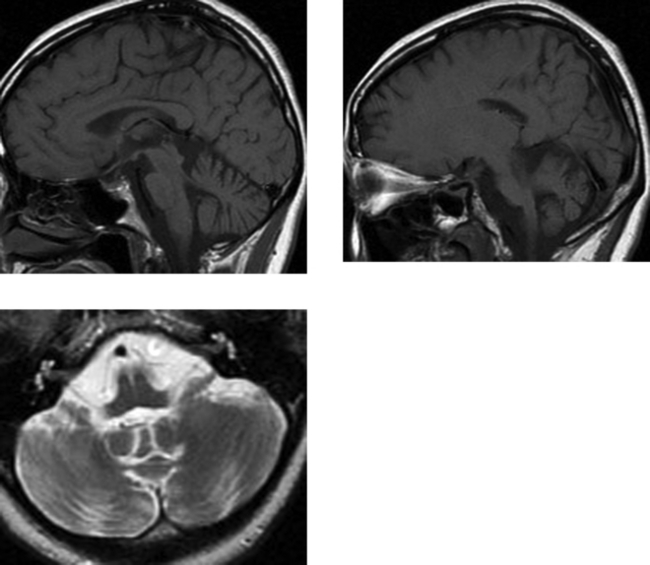
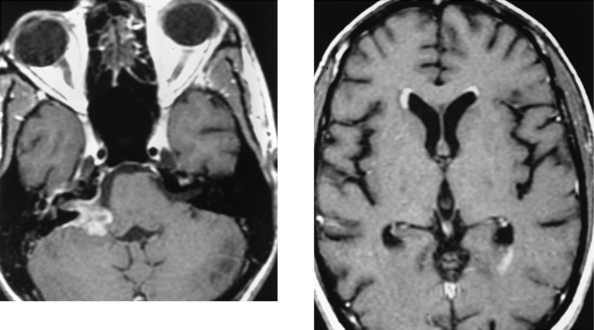
CASE 152 Olivopontocerebellar Degeneration
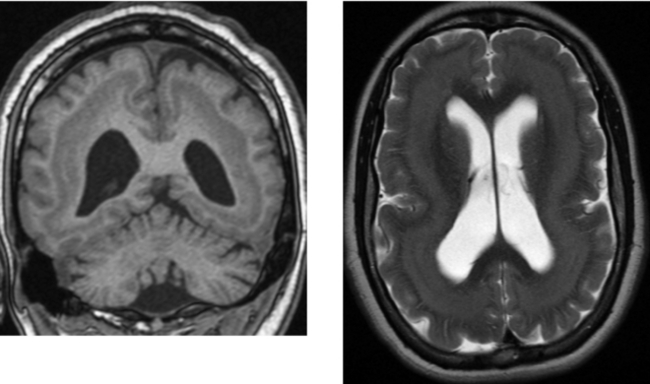
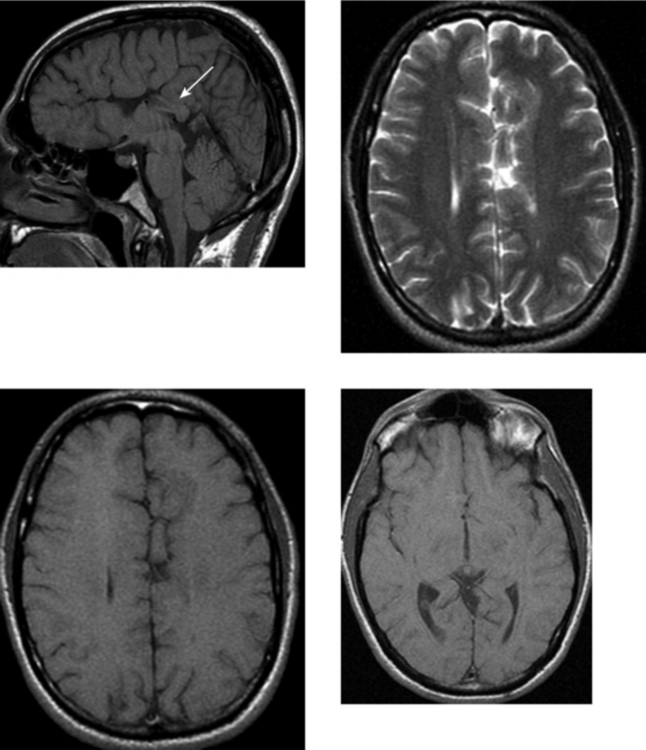
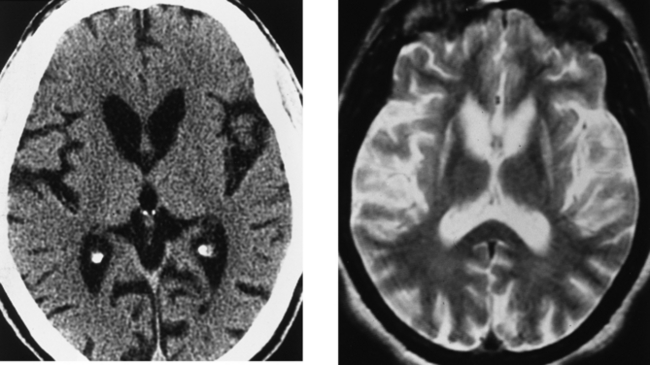
CASE 154 Huntington’s Disease
Krausz et al. 1996 Krausz Y, Bonne O, Marciano R, Yaffe S, Lerer B, Chisin R. Brain SPECT imaging of neuropsychiatric disorders. Eur J Radiol. 1996;21:183-187.
Montoya et al. 2006 Montoya A, Price BH, Menear M, Lepage M. Brain imaging and cognitive dysfunctions in Huntington’s disease. J Psychiatry Neurosci. 2006;31:21-29.
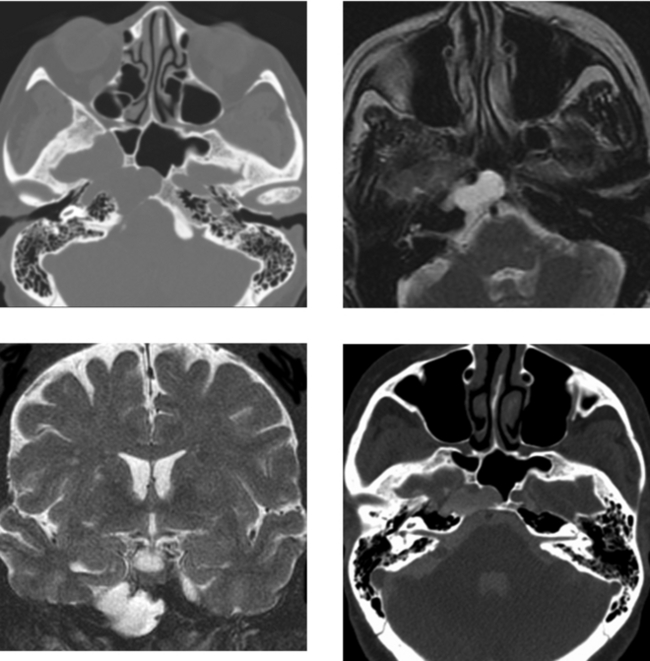
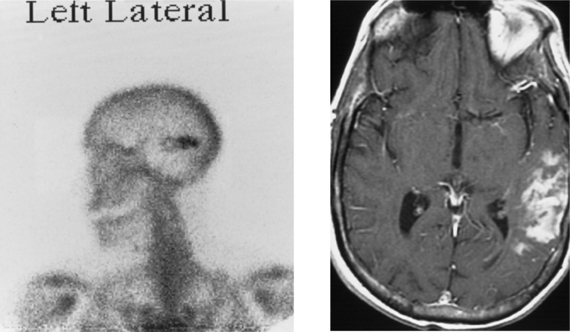
CASE 155 Meningocele and Pseudomeningocele of the Skull Base (Petrous Apex)
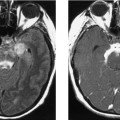
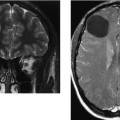
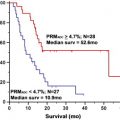
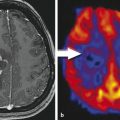
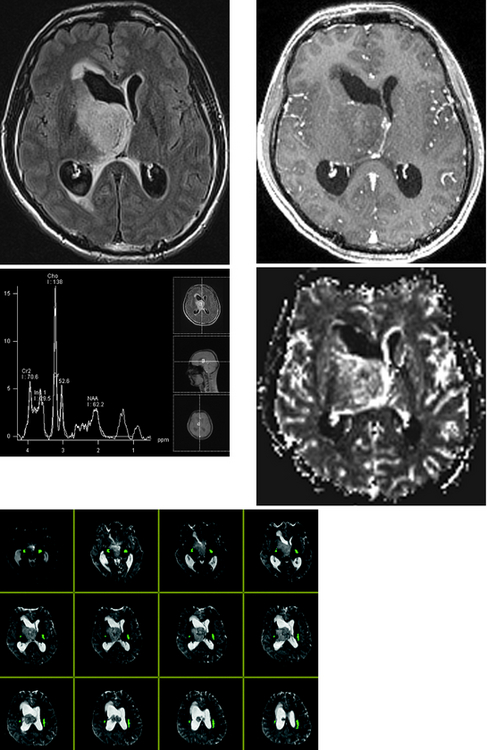
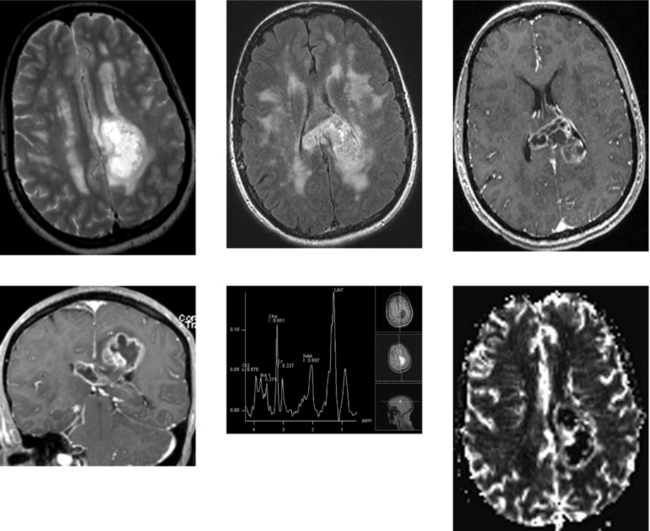
CASE 156 Right Thalamic Glioblastoma
Earnest FIV, Kelly PJ, Scheithauer BW, et al. Cerebral astrocytomas: histopathologic correlation of MR and CT contrast enhancement with stereotactic biopsy. Radiology. 1988;166:823-827.
Lupo JM, Cha S, Chang SM, Nelson SJ. Analysis of metabolic indices in regions of abnormal perfusion in patients with high grade glioma. AJNR Am J Neuroradiol. 2007;28:1455-1461.
CASE 157 Multiple Sclerosis and Glioblastoma
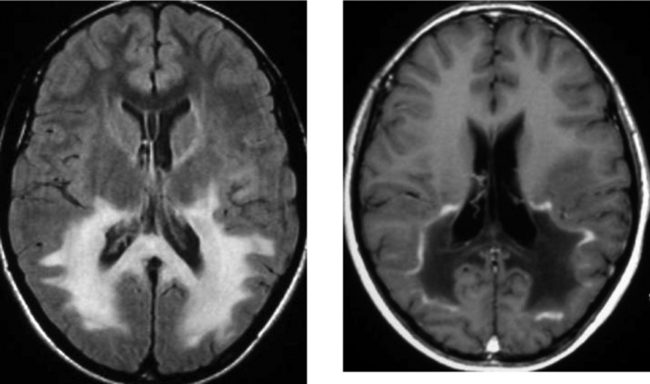
CASE 158 Adrenoleukodystrophy
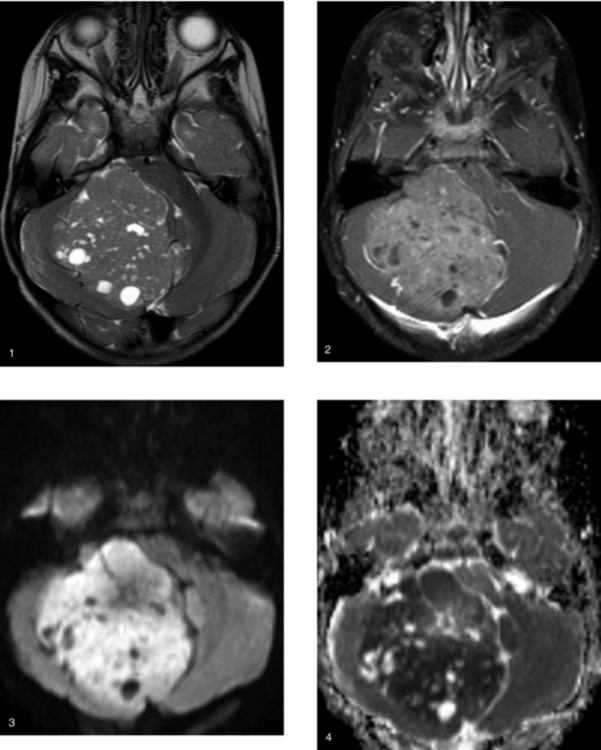
CASE 159 Medulloblastoma
CASE 160 Middle Cerebral Artery Territory Stroke Mimicking a Bone Metastasis
CASE 161 Cytomegalovirus Meningitis and Ependymitis in a Patient with AIDS
Boska MD, Mosley RL, Nawab M, et al. Advances in neuroimaging for HIV-1 associated neurological dysfunction: clues to diagnosis. pathogenesis and therapeutic monitoring, Curr HIV Res. 2004;2:61-68.
Vinters HV, Kwok MK, Ho HW, et al. Cytomegalovirus in the nervous system of patients with the acquired immune deficiency syndrome. Brain. 1989;112:245-268.
CASE 162 Upper Brainstem and Thalamic Cavernous Malformation
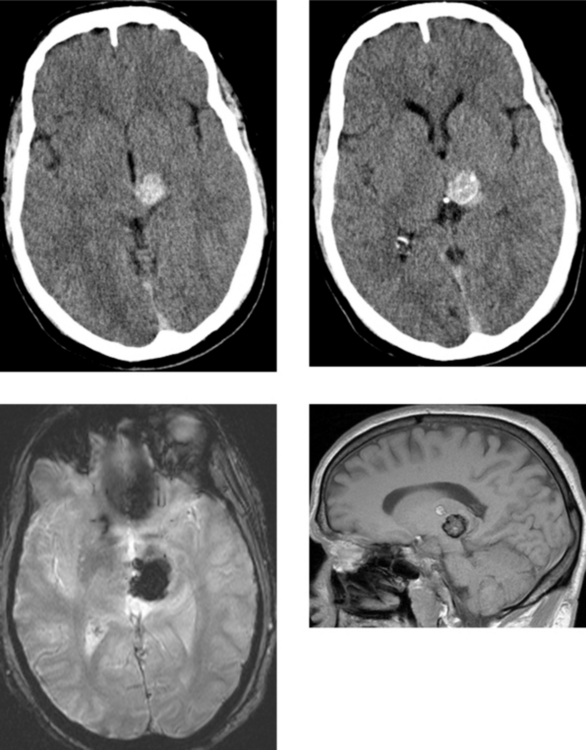
Porter RW, Detwiler PW, Spetzler RF, et al. Cavernous malformations of the brainstem: experience with 100 patients. J Neurosurg. 1999;90:50-58.
Zausinger S, Yousry I, Brueckmann H, Schmid-Elsaesser R, Tonn JC. Cavernous malformations of the brainstem: three-dimensional-constructive interference in steady-state magnetic resonance imaging for improvement of surgical approach and clinical results. Neurosurgery. 2006;58:322-330.
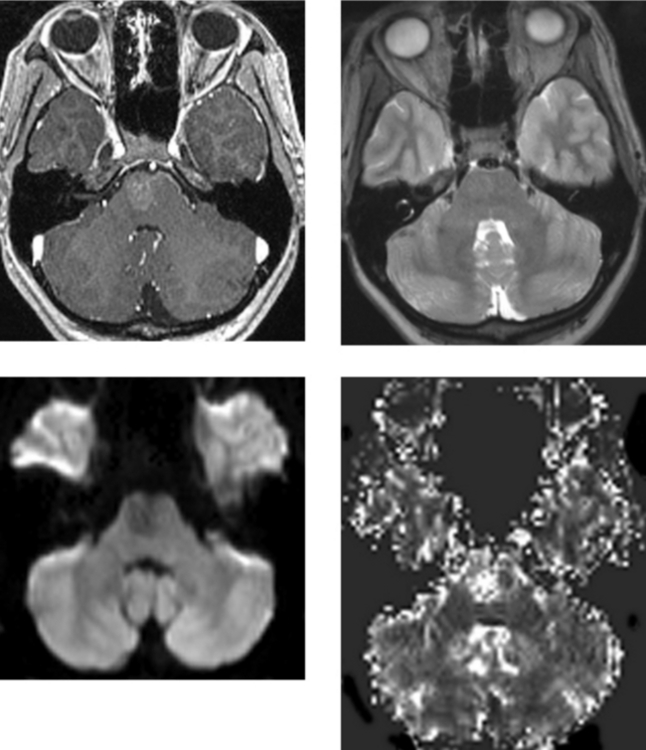
CASE 163 Capillary Telangiectasia and Developmental Venous Anomaly of the Brainstem
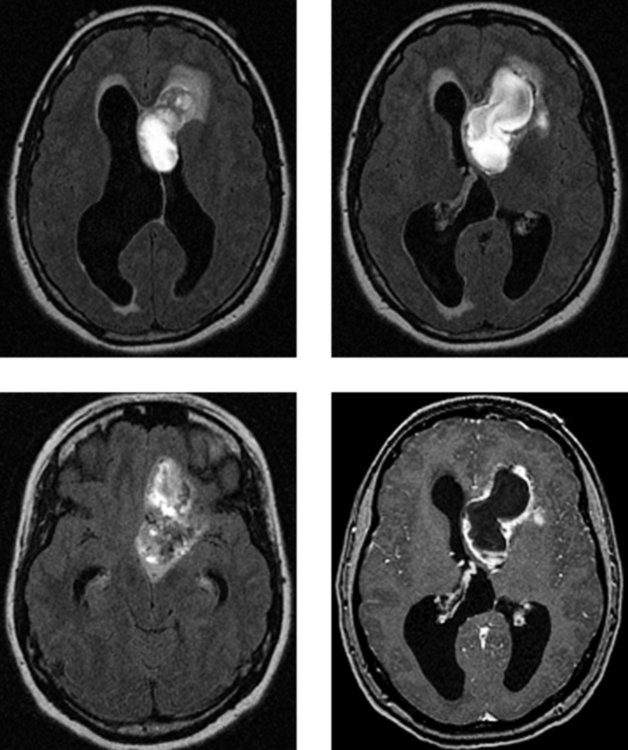
CASE 164 Central Neurocytoma
CASE 165 Marchiafava-Bignami Disease
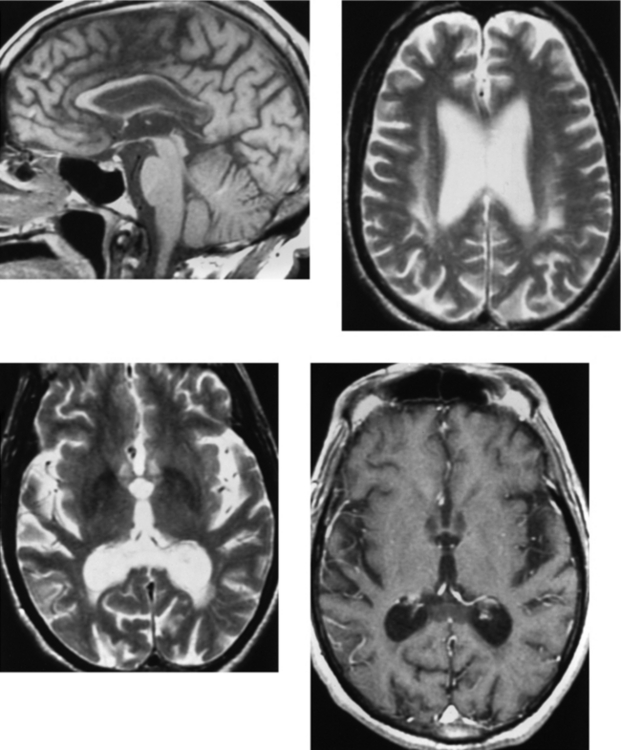
Arbelaez A, Pajon A, Castillo M. Acute Marchiafava-Bignami disease: MR findings in two patients. AJNR Am J Neuroradiol. 2003;24:1955-1957.
Kawarabuki K, Sakakibara T, Hirai M, Yoshioko Y, Yamamoto Y, Yamaki T. Marchiafava-Bignami disease: magnetic resonance imaging findings in corpus callosum and subcortical white matter. Eur J Radiol. 2003;48:175-177.
CASE 166 Metastatic Carcinoid to the Orbital Extraocular Muscles
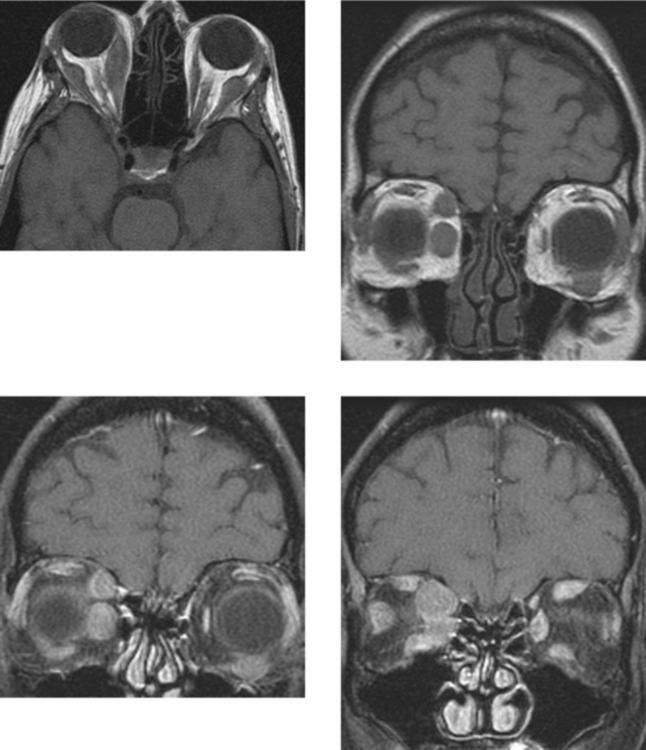
Borota OC, Kloster R, Lindal S. Carcinoid tumour metastatic to the orbit with infiltration to the extraocular orbital muscle. APMIS. 2005;113:135-139.
Hanson MW, Schneider AM, Enterline DS, Feldman JM, Gockerman JP. Iodine-131-mataiodobenzylguanidine uptake in metastatic carcinoid tumor to the orbit. J Nucl Med. 1998;39:647-650.
CASE 167 Joubert’s Syndrome