CHAPTER 49 Complex Congenital Heart Disease
Complex congenital heart disease is obviously one of the most challenging issues faced by the health care provider who takes care of the pediatric patient with cardiac lesions. Taken as a group, this set of lesions represents only a small portion of congenital heart disease (CHD) and is rare in the population; nevertheless, these diseases take up a considerable amount of the physician’s time. Hoffman and Kaplan1 have reported the results of a meta-analysis of the literature and found the incidence of moderate to severe forms of congenital heart disease to be 6/1,000 live births, rising to 19/1,000 live births if potentially serious bicuspid aortic valves are included. Putting this in perspective, all forms of CHD represent 75/1,000 live births, including such lesions as tiny muscular ventricular septal defects. In addition, the New England Infant Cardiac Program2 has reported that 3/1,000 live births need cardiac catheterization or surgery, or will die with CHD in early infancy (excluding premature infants with patent ductus arteriosus). This number rises to 5/1,000 live births who will need some type of specialized care during their lifetime. All these issues are a measure of the severity of the lesion. With improvements in diagnosis and treatment of CHD, along with a greater understanding of the anatomy and physiology, patients are living longer3 and represent a growing patient base seen by adult cardiologists and internists. In 1980, there were an estimated 300,000 adults with CHD, whereas this rose to approximately 1 million in 2000. In 2020, the number is anticipated to be 1.4 million.
TRANSPOSITION OF THE GREAT ARTERIES
Definition
The classic transposition of the great arteries (TGA) consists of isolated ventriculoarterial discordance, with the aorta arising from the right ventricle and the main pulmonary artery arising from the left ventricle.1,2,3 This results in separation of the systemic and pulmonary circulations, with limited mixing of oxygenated and deoxygenated blood. It usually refers to dextro-TGA (d-TGA; segmental anatomy {S,D,D}), in which the aortic valve is positioned anterior and to the right of the pulmonary valve. However, some refer to any segmental anatomy that results in this physiology as transposition. Occasionally, the conotruncus is further rotated so that the aorta is anterior and to the left of the pulmonary valve (segments {S,D,L}).
Prevalence and Epidemiology
The incidence of transposition of the great arteries is approximately 3 in 10,000 live births. It represents approximately 5% of all newborns with congenital heart disease.1 It is rarely associated with syndromes or extracardiac malformations, but is more common in infants of diabetic mothers.
Manifestations of Disease
Imaging Techniques and Findings
Radiography
The classic radiographic findings of transposition of the great arteries include a narrow superior mediastinum (so-called egg on a string) caused by the anteroposterior orientation of the great vessels and inapparent thymus, increased pulmonary vascular markings, and cardiomegaly. However, these findings are rarely diagnostic and, in fact, were shown to be absent in the vast majority of newborns with d-TGA (Fig. 49-1).4
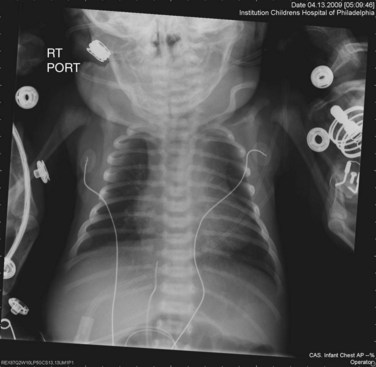
 FIGURE 49-1
FIGURE 49-1Ultrasound
Echocardiography can generally readily identify all the pertinent anatomic findings to manage patients with transposition of the great arteries. Subcostal imaging can readily identify the pulmonary artery arising from the left ventricle and the aorta arising from the right ventricle. The aorta can usually be identified by the coronaries and by its long length before branching (Fig. 49-2). The pulmonary artery branches early and the ductal arch should not be mistaken for the aorta (Fig. 49-3).
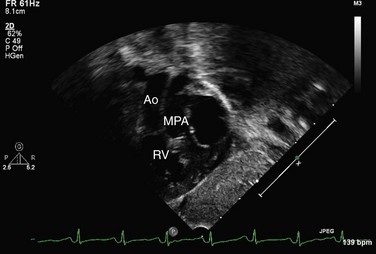
 FIGURE 49-2
FIGURE 49-2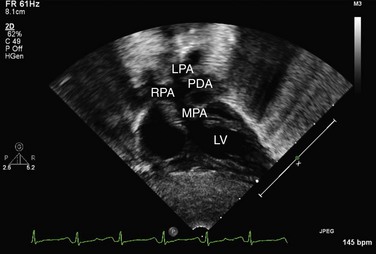
 FIGURE 49-3
FIGURE 49-3It is important to assess the atrial septum to determine whether it is restrictive (Fig. 49-4). In addition, the ventricular septum should be assessed to determine whether there are ventricular septal defects (Fig. 49-5). This is important for both preoperative management and surgical planning.
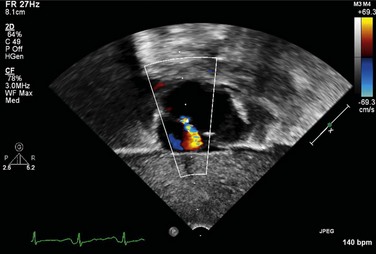
 FIGURE 49-4
FIGURE 49-4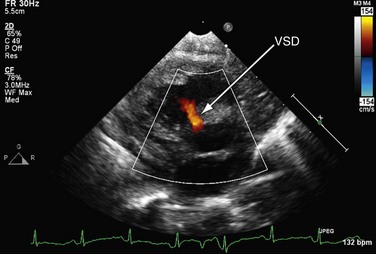
 FIGURE 49-5
FIGURE 49-5Magnetic Resonance Imaging
MRI plays an important role in the postoperative management of TGA. The standard for the treatment of TGA is the arterial switch (Jatene procedure), in which the aorta and main pulmonary artery are transected and sewn to the pulmonary and aortic root, respectively. The pulmonary arteries are generally brought anteriorly and draped over the aorta. The coronary arteries are transferred separately with buttons of tissue from the aorta to avoid ostial stenosis. Several problems can result, for which cardiac MRI is well suited to investigate. Supravalvular stenosis can occur at either anastomosis site (Fig. 49-6). Cine and velocity mapping can be used effectively to define regions of stenosis and quantify the degree of acceleration. Three-dimensional gadolinium sequences can also be useful to define stenoses and their relationships to other structures (Fig. 49-7). Unilateral or occasionally bilateral branch pulmonary artery stenosis may occur from stretching the branch pulmonary arteries after the Lecompte maneuver. Insufficiency of either valve can result from distortion during surgery. Through-plane velocity mapping can be used to quantify the degree of insufficiency of the valves precisely. Furthermore, short-axis cine volume sets can quantify the ventricular size, ejection, and wall motion abnormalities to screen for the effects of valve regurgitation or coronary abnormalities. Whole heart sequences can be used to evaluate for ostial stenosis of the transferred coronaries (Fig. 49-8). Perhaps more importantly, MRI can be used to evaluate for perfusion defects secondary to coronary stenosis. Gadolinium perfusion imaging is generally performed at rest and during adenosine administration. Adenosine administration causes coronary vasodilation and accentuates perfusion abnormalities by “stealing” flow from regions of marginal coronary perfusion.
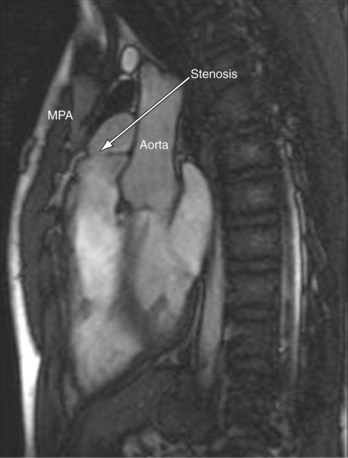
 FIGURE 49-6
FIGURE 49-6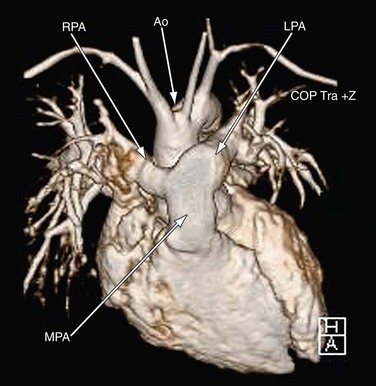
 FIGURE 49-7
FIGURE 49-7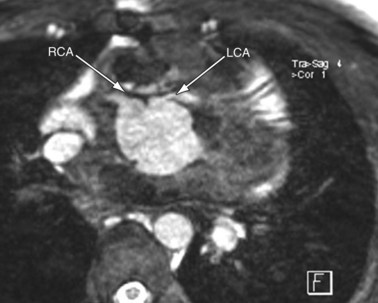
 FIGURE 49-8
FIGURE 49-8See later, “Single Ventricle,” for a more detailed description of the routine cardiac MRI examination.
TETRALOGY OF FALLOT
Prevalence and Epidemiology
The incidence of tetralogy of Fallot is approximately 4 in 10,000 live births, accounting for approximately 7% to 10% of cases of congenital heart disease.5 It occurs equally in males and females, and represents one of the most common lesions requiring intervention in the first year of life. It occurs commonly in association with genetic defects, including Down syndrome, DiGeorge syndrome (22q11 microdeletion), and Alagille syndrome (Jag1 mutation).6,7
Etiology and Pathophysiology
While originally described by Fallot as a constellation of four findings,8 it is now understood that the pathogenesis appears to be related to a single abnormality. The anterior malalignment VSD with normally related great vessels is responsible for the associated aortic override and right ventricular outflow tract obstruction, which classically results in right ventricular hypertrophy.
Certain associated conditions may be important to the management of tetralogy of Fallot. A right aortic arch occurs in approximately 25% of patients. Coronary anomalies are common (approximately 9%), including the left anterior descending (LAD) artery from the right coronary and single coronary.9 Pulmonary atresia may occur, with or without the presence of major aortopulmonary collaterals. Patients may often have additional muscular VSDs, and it may occur in association with a complete common atrioventricular canal, especially in association with Down syndrome. Stenosis of the left pulmonary artery is common and, rarely, isolation of the pulmonary artery contralateral to the aortic arch may occur. A patent ductus arteriosus is common; when there is significant outflow tract obstruction, the duct may be tortuous.
Manifestations of Disease
Imaging Techniques and Findings
Radiography
The classic radiographic finding is a boot-shaped heart, with an upturned apex (Fig. 49-9). Other findings may range from a normal heart size and decreased pulmonary blood flow to an increased heart size and increased pulmonary blood flow, depending on the degree of obstruction. A right aortic arch can be noted on the radiograph.
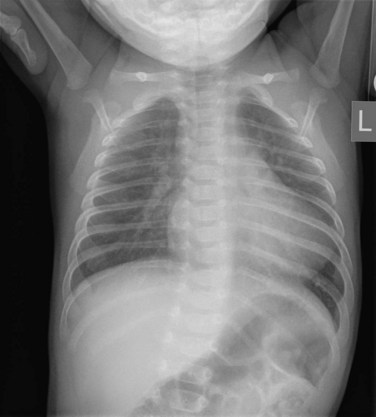
 FIGURE 49-9
FIGURE 49-9Ultrasound
The mainstay of the preoperative evaluation of tetralogy of Fallot is echocardiography. In addition to making the diagnosis (Fig. 49-10), the echocardiogram should focus on the degree and location (subpulmonary, valvular or supravalvular) of right ventricular outflow obstruction (Fig. 49-11), the presence of additional ventricular septal defects, the size and origin of the branch pulmonary arteries, the presence of aortopulmonary collaterals, the coronary origins and courses (with particular attention to whether the LAD or other major branch crosses the right ventricular outflow tract [RVOT]), the arch sidedness and branching pattern, and the patency and course of the ductus arteriosus. With rare exception, these can be evaluated by routine transthoracic imaging.
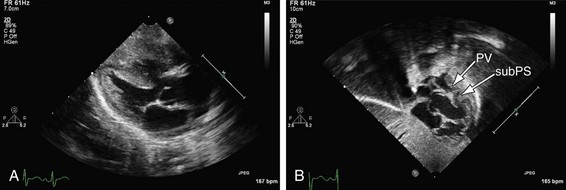
 FIGURE 49-10
FIGURE 49-10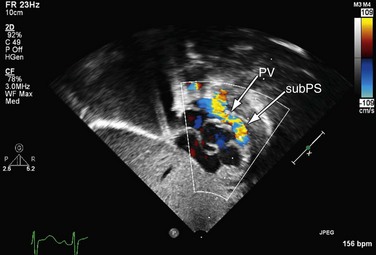
 FIGURE 49-11
FIGURE 49-11Magnetic Resonance Imaging
The preoperative use of cardiac MRI is limited to specific situations. In some cases, it may be useful to use respiratory and cardiac-gated, T2-prepared whole heart imaging to define the origins of the coronary arteries if they are not well seen by echocardiography. Gadolinium angiography is also useful to define aortopulmonary collaterals, and has been shown to be as effective as traditional angiography.10
MRI has become an important part of the postoperative management of tetralogy of Fallot. The repair for tetralogy of Fallot generally involves patch closure of the VSD and relief of the RVOT obstruction. Some patients may have an adequate pulmonary valve annulus and require only resection of an RV muscle bundle and/or pulmonary valvotomy. However, many will require a transannular patch. Patients in which a major coronary branch crosses the RVOT may require an RV to PA conduit, creating a double-barreled outflow. In patients who have had a transannular patch as part of the repair, pulmonary insufficiency may cause progressive RV dilation and decreased RV performance. In addition, left ventricular performance may decline, likely secondary to interactions with the left ventricle. Cardiac MRI can quantify the pulmonary regurgitation and right ventricular size, and is important in the monitoring of patients who have evidence of significant RV dilation by echocardiography or have poor echocardiographic windows (Figs. 49-12 and 49-13). MRI can also effectively evaluate residual RVOT obstruction or conduit stenosis, branch pulmonary artery stenosis, and pulmonary flow distribution to each lung (Fig. 49-14).
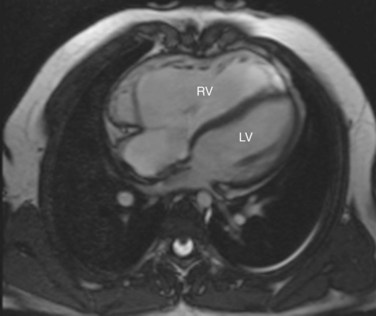
 FIGURE 49-12
FIGURE 49-12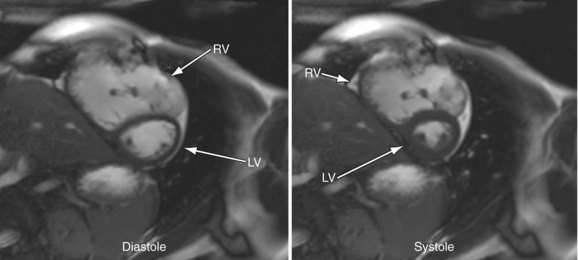
 FIGURE 49-13
FIGURE 49-13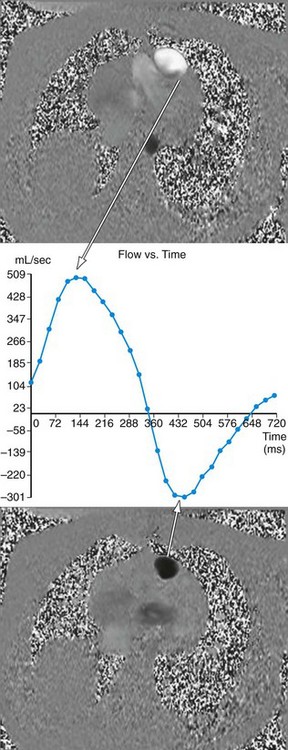
 FIGURE 49-14
FIGURE 49-14See later, “Single Ventricle,” for a more detailed description of the routine cardiac MRI examination.
Nuclear Medicine: Positron Emission Tomography
Nuclear scintigraphy perfusion imaging has been used postoperatively in patients with TOF in the setting of branch pulmonary artery stenosis to quantify pulmonary blood flow distribution.11 However, this has largely been supplanted by cardiac MRI velocity mapping.12,13 It still may be useful for patients in whom a stent or coil artifact precludes assessment by MRI.
TRUNCUS ARTERIOSUS
Definition
There are two major classification schemes in use—those of Collett and Edwards14 and Van Praagh.15 These are invariably based on the position of the main pulmonary artery segment and branch pulmonary arteries, with the Van Praagh classification using types A and B to delineate whether a VSD is present (almost all have VSDs). Of the different forms, 92% of all patients fall into type 1A or 2A. The following are the definitions used in the Van Praagh classification with its differences and similarities with the Collett and Edwards classification noted; the Van Praagh classification takes into account aortic arch anomalies.
Prevalence and Epidemiology
According to Hoffman and Kaplan,1 the mean incidence of truncus arteriosus is 107 per million live births; it is thought to occur in 1% to 2% of patients with CHD at necropsy and represents approximately 0.7% of all congenital heart disease. The DiGeorge syndrome and patients with microdeletion of chromosome 22 have a high incidence of having truncus arteriosus. There is no race or gender predilection.
Etiology and Pathophysiology
Etiology
This rare lesion is caused by a failure in conotruncal septation of the embryonic truncus arteriosus and conus. The truncus is normally divided into the aorta and pulmonary artery by two ridges that appear in the 4- to 5-week-old embryo and that grow toward the midline. Cells from the neural crest directly contribute to this septation; one of the leading theories is that this malformation is a disruption in neural crest migration. Van Praagh and Van Praagh, however, believed this to be a form of tetralogy of Fallot.15
Pathophysiology
There are associated cardiovascular malformations, such as abnormalities of the coronary arteries, a right aortic arch, persistent left superior vena cava, aberrant origin of the left subclavian, patent foramen ovale, partial and complete atrioventricular canal defects, mitral and tricuspid malformations, double-inlet or hypoplastic left ventricle, left pulmonary artery sling, and anomalous pulmonary venous connections.16
Manifestations of Disease
Imaging Techniques and Findings
Ultrasound
Postoperatively, narrowing of the reconstructed right ventricular outflow tract and pulmonary arteries needs to be assessed. Stenosis can be evaluated by color flow mapping and Doppler echocardiography can determine the gradient; this is best performed in the subcostal sagittal or parasternal short-axis views and, occasionally, in the apical view angled extremely anteriorly. Residual ventricular septal defects can be seen by short-axis sweeps. Assessment of the truncal valve, as in the preoperative assessment, must be made routinely (Figs 49-15 and 49-16).
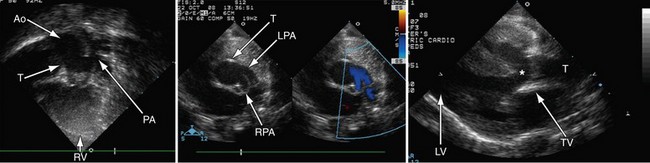
 FIGURE 49-15
FIGURE 49-15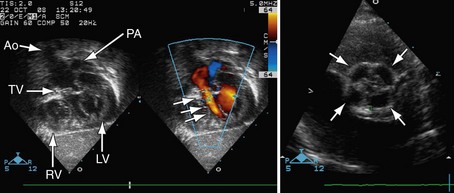
 FIGURE 49-16
FIGURE 49-16Computed Tomography
Because of radiation considerations, CT scanning plays a limited role in the care of the patient with congenital heart disease, and truncus arteriosus is no different. It is done chiefly when there is a contraindication to MRI and, when used, it is generally carried out postoperatively to visualize the right ventricular outflow tract and branch pulmonary arteries along with the aortic reconstruction if the aortic arch was interrupted. Because of its very limited temporal resolution, except if absolutely needed, ventricular function is best determined by MRI or echocardiography (Figs. 49-17 to 49-19).
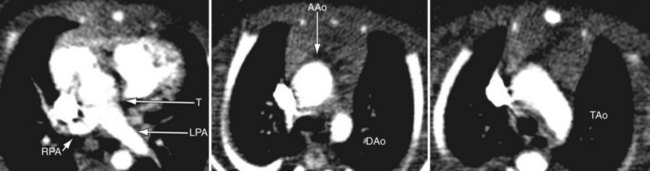
 FIGURE 49-17
FIGURE 49-17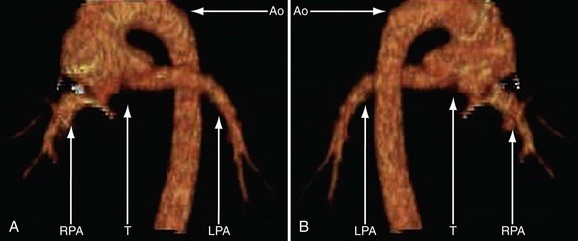
 FIGURE 49-18
FIGURE 49-18Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


