Complex Cyanotic Congenital Heart Disease
Teresa Chapman, MD
Randolph K. Otto, MD
LEARNING OBJECTIVES
1. Provide an appropriate differential diagnosis for congestive heart failure in the newborn.
2. Name one congenital heart lesion that explains radiographic pulmonary oligemia with heart enlargement, and name one without heart enlargement.
3. Explain the abnormalities underlying the “boot-shaped” appearance of the cardiac silhouette in tetralogy of Fallot.
4. Describe the three-stage surgical approach to hypoplastic left heart syndrome (HLHS).
5. Name three MRI findings that would predict failure of a biventricular repair for HLHS.
6. Describe why congenitally corrected transposition of the great arteries (l-TGA) is not so “correct.”
7. Predict whether or not TGA is present in the setting of tricuspid atresia based on pulmonary blood flow.
8. List the types of TAPVR may present with venous congestion secondary to obstruction.
INTRODUCTION
Complex congenital heart disease (CHD) encompasses a number of life-threatening cardiac malformations in the newborn by causing systemic arterial oxygen desaturation. Complex CHD has a prevalence of 1.45 per 1,000 children.1 Early recognition of these malformations is possible in the fetus with progressively improving capabilities of ultrasound and magnetic resonance imaging, allowing for appropriate delivery planning and immediate multispecialist care for neonates with diagnosed complex CHD. Nevertheless, a substantial number of cases are diagnosed postnatally by clinical signs and symptoms, including tachypnea and central cyanosis, which refers to a bluish discoloration of the tissues resulting from an absolute reduction in hemoglobin of 3 g/dL or more in the capillary bed.2
The imaging evaluation of these patients goes far beyond establishing the primary diagnosis—ever-improving treatments for CHD allow for many neonates to survive well into adulthood. Surgical palliative and corrective procedures, often performed in a staged approach over time, require repeated follow-up imaging studies to evaluate for possible complications of CHD and surgery.3,4 In this chapter, the advantages of various imaging modalities are discussed, in addition to specific details about the more common and important complex and cyanotic CHDs. The fundamental concepts regarding surgical reparative procedures are reviewed as well.
IMAGING MODALITIES
Chest radiography is the appropriate initial imaging modality utilized for a clinically suspected cardiopulmonary abnormality. This modality has been shown to be a good initial diagnostic tool for detecting significantly abnormal pulmonary vascularity in children with CHD.5 As discussed in Chapters 9 and 10, assessment of overall cardiac size, chamber enlargement, and pulmonary blood flow offers clues to the etiology of the infant’s distress. If the infant is cyanotic, and the pulmonary blood flow appears diffusely decreased (oligemic), this implies a right heart or right ventricular outflow tract obstructive lesion. In other words, circulating blood volume is unable to reach the pulmonary arteries for oxygenation because of a structural abnormality, classified by Higgins as a group 3 lesion (Table 10.1).6 The general findings of increased cardiac silhouette size and increased pulmonary blood flow in a cyanotic newborn should prompt consideration of an admixture type of CHD (the group 4 lesions, see Table 10.1). Admixture lesions include the following: transposition of the great vessels, tricuspid atresia, truncus arteriosus, total anomalous pulmonary venous return, and single-ventricle pathophysiology. In the noncyanotic infant, however, findings of a large heart size and increased pulmonary blood flow could also signify either an intracardiac shunt (discussed in Chapter 10) or congestive heart failure. Potential etiologies for congestive heart failure in an infant are summarized in Table 11.1. It is critical to be aware that one of the most common causes of congestive heart failure within the first 24 hours of life is hypoplastic left heart syndrome (HLHS),7 which is discussed later in this chapter.
Advantages of the various cardiac imaging techniques are discussed in Chapter 9 and are briefly summarized here. Electrocardiography (ECG)-gated and respiratory-gated crosssectional imaging by CT and MR are continually evolving
modalities that are used both prior to surgical repair and following surgery to monitor ventricular function and surgical conduit patency. With multidetector CT, prospective ECG gating reduces the amount of ionizing radiation needed by replacing a spiral technique with a stepwise, sequential technique, such that the effective dose is essentially the same as for non-ECGgated MDCT.8 As the use of CT has risen, the number of conventional angiography cases for cardiac assessment has decreased, which offers a further reduction in exposure to ionizing radiation (median-estimated effective doses for cardiac CT using a retrospective technique have been published at 1.7 mSv versus diagnostic angiography at 5.0 mSv).9 MR imaging has the disadvantage of longer acquisition times than CT angiography but offers the advantages of improved temporal resolution and the absence of ionizing radiation. The sequences in cardiac MR can be optimized to evaluate cardiac and vascular morphology, function, and flow.10
modalities that are used both prior to surgical repair and following surgery to monitor ventricular function and surgical conduit patency. With multidetector CT, prospective ECG gating reduces the amount of ionizing radiation needed by replacing a spiral technique with a stepwise, sequential technique, such that the effective dose is essentially the same as for non-ECGgated MDCT.8 As the use of CT has risen, the number of conventional angiography cases for cardiac assessment has decreased, which offers a further reduction in exposure to ionizing radiation (median-estimated effective doses for cardiac CT using a retrospective technique have been published at 1.7 mSv versus diagnostic angiography at 5.0 mSv).9 MR imaging has the disadvantage of longer acquisition times than CT angiography but offers the advantages of improved temporal resolution and the absence of ionizing radiation. The sequences in cardiac MR can be optimized to evaluate cardiac and vascular morphology, function, and flow.10
Table 11.1 POSSIBLE CAUSES OF CONGESTIVE HEART FAILURE IN A NEWBORN, OTHER THAN COMPLEX CHD | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||
CONGENITAL CARDIAC MALFORMATIONS WITH RIGHT VENTRICULAR OUTFLOW TRACT OBSTRUCTION
An essential concept pertaining to right heart malformations is the decrease in blood flow through the right heart, with resulting central cyanosis and pulmonary oligemia. This group of lesions can be divided further into those with cardiomegaly (Group 3 in Table 10.1, Ebstein anomaly being the classic example) and those without cardiomegaly (Group 2, most commonly tetralogy of Fallot).
EBSTEIN ANOMALY
Ebstein anomaly is a rare cardiac malformation of the tricuspid valve, accounting for less than 1% of all CHD cases.11 In this disorder, the septal and posterior leaflets of the tricuspid valve are dysplastic and downwardly displaced. The result is marked dilatation of the right atrium, “atrialization” of the right ventricle, and obstruction of deoxygenated blood flow to the lungs (Fig. 11.1A). Cardiac MR imaging may be used to assess the right ventricular function and the morphology of the tricuspid valve (Fig. 11.1B, C). Surgical repair results are best if the tricuspid valve anterior leaflet is mobilized and the atrialized chamber is plicated (folded), avoiding the need for valve prosthesis.12
TETRALOGY OF FALLOT
Tetralogy of Fallot (TOF) is the most common type of cyanotic CHD, with an incidence of 0.28 per 1,000 live births, equally affecting males and females.13 The likelihood of a genetic substrate to TOF is based on a recognized association with a chromosome 22 microdeletion; TOF is frequently diagnosed in those with DiGeorge syndrome or velocardiofacial syndrome, both of which have 22q11 deletions.14,15 Arising from the embryologic defect of an anteriorly and superiorly deviated conal septum is the tetrad of ventricular septal defect, overriding aorta, right ventricular outflow obstruction, and right ventricular hypertrophy.10,13 The pentalogy of Fallot includes an atrial septal defect, which is seen in 10% of cases.4 Other associated cardiac anomalies include a right-sided aortic arch with mirror-image branching (seen in 25% of TOF cases), coronary artery anomalies (seen in 10%), and peripheral pulmonary arterial stenoses.
Before the development of surgical intervention for TOF, about half of affected patients would die within the first few years of life, whereas today, virtually all individuals born with this malformation can survive to adulthood with surgical repair. Most centers prefer to operate on infants of 3 to 6 months, unless severe cyanosis merits surgery sooner.13,16 Early surgical palliation involves closure of the VSD and enlargement of the right ventricular outflow tract, occasionally utilizing a pericardial patch. This generally results in pulmonary regurgitation that will eventually lead to right ventricular dilation and failure. Pulmonary valve replacement is often required later in childhood to prevent irreversible right ventricular failure.
Imaging by radiography may show the following characteristics: (1) elevation of the cardiac apex above the left hemidiaphragm due to right ventricular hypertrophy; (2) absence of the expected main pulmonary artery shadow—this creates a concave or scooped appearance at the aortopulmonary window and, together with the RV hypertrophy, leads to the cardiac silhouette resembling a boot, hence referral to a “boot-shaped heart”; (3) diminished pulmonary flow; and (4) a right-sided aortic arch in one-quarter of cases (Fig. 11.2A). Preoperative assessment of TOF with cardiac MR angiography is usually reserved for those with complex pulmonary arterial anatomy and major aortopulmonary collateral vessels (MAPCAs).10 Postoperative assessment is a more common indication for evaluation of TOF patients by cardiac MR to identify complications, including pulmonary regurgitation, outflow tract restenosis, aneurysm, residual VSD, and branch pulmonary artery stenosis (Fig. 11.2B). Current guidelines for pulmonic valve replacement include (1) the onset of clinical symptoms, (2) progressive tricuspid valve regurgitation, and (3) progressive RV dilation on imaging. Cardiac MR is most often employed to measure right ventricular end-diastolic volume (RVEDV), and different centers have variable thresholds for RVEDV prompting pulmonary valve replacement.17
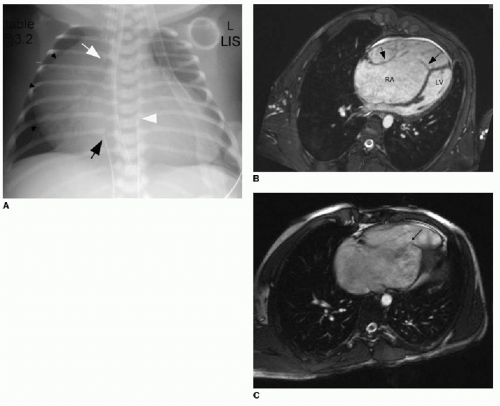 FIG. 11.1 • Ebstein anomaly. Chest radiograph (A) of a neonate shows marked cardiomegaly, predominantly related to marked right atrial enlargement (black arrowheads). Endotracheal tube (white arrow), umbilical arterial (white arrowhead), and umbilical venous catheter (black arrow) are present. B: Cardiac MRI on a different patient (multiplanar rendering of steady-state free precession sequence) shows atrialization of the right ventricle and apical displacement of the tricuspid valve leaflets (arrows) (RA, right atrium; LV, left ventricle). Tricuspid valve regurgitation is evident (C), based on dark flow jet across the valve during systole (small black arrow). |
CONGENITAL CARDIAC ADMIXTURE MALFORMATIONS
Complete mixing of pulmonary and systemic venous return within the cardiac chambers or great artery level is referred to as admixture physiology. The essential concept to understand about the admixture lesions is that these infants are born with central cyanosis. Common radiographic features include prominent cardiac silhouette and increased pulmonary blood flow (group 4 lesions, see Table 10.1).
HYPOPLASTIC LEFT HEART SYNDROME
The pathophysiologic concepts and treatment approaches for HLHS can be extrapolated to a wider understanding of singleventricle congenital cardiac malformations, of which HLHS is the most common. HLHS has a prevalence of approximately 0.2 per 1,000 live births18,19 and occurs more frequently in boys.20 Without treatment, the malformation is invariably lethal.21,22 HLHS includes a wide several developmental abnormalities affecting the left side of the heart, including hypoplasia of the left ventricle, mitral valve, aortic valve, and ascending aorta (see Fig. 11.3A for a schematic diagram). It is hypothesized that the embryologic development of this set of malformations is a stepwise cascade of miscues.7
At birth, patency of the ductus arteriosus must be maintained postnatally by administering prostaglandin E1 to preserve prenatal hemodynamics for survival as a bridge to surgical correction.18 In patients with HLHS, flow through the patent ductus arteriosus is right to left during ventricular systole, providing flow to the systemic circulation. During ventricular diastole, there is leftto-right flow across the patent ductus arteriosus and retrograde flow through the ascending aorta to perfuse the coronary arteries. Severe congestive heart failure ensues when the left atrial blood volume cannot enter the left ventricle and outflow tract. Surgical management of HLHS infants involves a three-stage approach (Norwood procedure).
The first stage involves creation of a “neoaorta” utilizing the main pulmonary artery to reconstruct the aorta, with the right ventricle thereby assuming a systemic role. The atrial septum is excised to allow for intra-atrial flow, and the ductus arteriosus is ligated. Initially, a modified Blalock-Taussig (BT) shunt (a synthetic conduit) was used to provide pulmonary arterial blood
flow (usually done on the right side from the innominate or subclavian arteries) to the right pulmonary artery to minimize volume load on the right ventricle and provide sufficient pulmonary arterial perfusion pressure in the neonatal period.18,22 Alternatively, a Sano shunt may be used to provide pulmonary arterial flow from the right ventricle, with less distortion of the pulmonary arteries. Frequently, a modification of the original Norwood procedure includes the Damus-Kaye-Stansel procedure, whereby the hypoplastic native ascending aorta is anastomosed to the neoaorta to allow perfusion of the coronary arteries. Once pulmonary arterial resistance has decreased to normal levels (usually at 3 to 6 months of age), stage two of the Norwood procedure involves the bidirectional Glenn procedure, a cavopulmonary shunt creation, to more completely relieve load off of the right ventricle and stimulate further growth of the pulmonary arteries. This procedure involves creation of an end-to-side anastomosis between the superior vena cava and the main pulmonary artery, followed by removal of the previously established systemic arterial to pulmonary shunt. Finally, the third palliative stage involves the Fontan completion




flow (usually done on the right side from the innominate or subclavian arteries) to the right pulmonary artery to minimize volume load on the right ventricle and provide sufficient pulmonary arterial perfusion pressure in the neonatal period.18,22 Alternatively, a Sano shunt may be used to provide pulmonary arterial flow from the right ventricle, with less distortion of the pulmonary arteries. Frequently, a modification of the original Norwood procedure includes the Damus-Kaye-Stansel procedure, whereby the hypoplastic native ascending aorta is anastomosed to the neoaorta to allow perfusion of the coronary arteries. Once pulmonary arterial resistance has decreased to normal levels (usually at 3 to 6 months of age), stage two of the Norwood procedure involves the bidirectional Glenn procedure, a cavopulmonary shunt creation, to more completely relieve load off of the right ventricle and stimulate further growth of the pulmonary arteries. This procedure involves creation of an end-to-side anastomosis between the superior vena cava and the main pulmonary artery, followed by removal of the previously established systemic arterial to pulmonary shunt. Finally, the third palliative stage involves the Fontan completion
Related posts:
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
