 Normal and Abnormal Embryogenesis of the Spine: An Overview
Normal and Abnormal Embryogenesis of the Spine: An Overviewpolarized cellular protrusions that enable the cells to migrate medially and intercalate with neighboring cells close to the midline (3). This midline convergence of cells causes an anteroposterior elongation and narrowing of the neural plate (4,5). This process is strongly related to the development of cell polarity (4,5). Along with the process of shaping, the neural plate also bends. At approximately 17 days of gestation, the lateral portions of the neural plate begin to thicken bilaterally, forming the neural folds; the process of bending elevates these folds and brings them to the dorsal midline (6). The process involves the formation of “hinge points” at two sites: the median hinge point (MHP) in the ventral midline and extending over the rostrocaudal extent of the neural plate and the paired dorsolateral hinge points (DLHPs), which form mainly at the levels of the developing brain (2) and lower spine. The formation of these hinge points is controlled by secretion of the signal transduction protein sonic hedgehog by the notochord as well as an inhibitory interaction between BMP2 and Noggin, particularly in the lower spine (7). After formation of the hinge points, the more lateral aspects of the neural plate are elevated around the MHP, bringing the DLHPs upward and toward the midline (Fig. 9-2). This elevation is accomplished by a poorly understood process called apical constriction, in which columnar cells of the neural tube are converted into wedge-shaped cells (8). Eventually, the lateral folds contact one another in the dorsal midline and adhere to one another, with their fusion forming the neural tube (neurulation). This midline contact (also called neural fold apposition) results from constriction of the open posterior neural tube, which is biomechanically coupled to the zippering point by an F-actin network (9). Neurulation seems to begin separately at least two different levels in humans, when cellular protrusions (possibly cilia) project medially from the most dorsal cells of the neural folds on either side. A third site of closure at the caudal end of the embryo has recently been identified in mouse embryos; if present in humans, this may account for the high incidence of spina bifida at this level (9). Cell recognition and adhesion occur under the influence of many molecules (Ephrin-A5, EphA7, neural cell adhesion molecule, and neural cadherin among them (3)), closing the tube at each point. Immediately following closure, the overlying ectoderm separates from the neural tissue and the edges of the ectoderm meet in the midline and fuse, forming a continuous ectodermal covering of the neural structures, with the mesenchymal cells of the neural crest migrating between the cutaneous and neural ectoderm layers (2). Progressive folding and closure of the neural structures and separation from ectoderm then proceed both cranially and caudally from each point of initial closure, ultimately resulting in complete closure (10,11). The initial closure of the neural tube in humans, the posterior neuropore, is believed to be at the hindbrain-cervical junction, from which closure extends in both directions. A second site of closure in mice is at the forebrain-midbrain boundary; this site has not been confirmed in humans. The necessity of this site even in mice is questionable, as about 80% of mice that lack the second closure point still achieve complete cranial closure (12). The “third” site of closure occurs at the most rostral extremity of the forebrain, the lamina terminalis (2). The exact site of the most caudal end of the neurulation-formed neural tube has been debated, but most experts believe that it is at the S2 level (10,11). Others point out, however, that neural tube defects are not restricted to any specific location(s) and propose that the human neural tube initially closes at a single site with closures extending from that location (13).
 Figure 9-1 Schematic diagram showing the development of the neural plate. The primitive streak forms along the surface of the embryo by about 15 days of embryonic life. A small primitive pit lies at what will become the cephalic end of the primitive streak; a nodular proliferation of cells surrounding the primitive pit becomes known as the Hensen node (A, seen from above and B, midline cut of A). On about days 15 and 16, cells enter the primitive pit and migrate cephalad in the midline to form the notochordal process, which will eventually become the notochord. The notochordal process and notochord induce formation of a plate of ectodermal cells dorsally in the midline; this is the neural plate (C, seen from above and D, midline cut of C). |
we review these cellular, molecular, and biomechanical mechanisms involved in neural tube closure, based on studies of various vertebrate species, focusing on the most recent advances in the field.
 Figure 9-2 Normal and abnormal neurulation. A-E. Normal neurulation. The neural plate is composed of neural ectoderm, which is continuous with cutaneous ectoderm on either side. The cells at the junction of the neural ectoderm and cutaneous ectoderm will eventually differentiate into neural crest cells (A). At approximately 17 days, the lateral portions of the neural plate begin to thicken, forming the neural folds (B). Contractile filaments located in the neuroepithelial cells in the neural folds contract, causing the neural folds to bend dorsally along the entire length of the neuroaxis, bringing the edges of the neural folds toward one another in the midline (C). Neurulation (closure of the neural tube) begins when the neural folds meet in the midline. At the time of closure, the overlying ectoderm separates from the neural tissue and fuses in the midline, forming a continuous ectodermal covering of the neural structures. At the same time, the neural crest cells are extruded from the neural tube to form a transient structure immediately dorsal to the tube (D). Eventually, these neural crest cells will migrate to form dorsal root ganglia and multiple other structures (E). F-G. Abnormal neurulation. When there is premature disjunction of neural ectoderm from cutaneous ectoderm, the surrounding mesenchyme gains access to the inner surface of the neural tube. When mesenchyme comes in contact with this primitive ependymal lining, it evolves into fat. This is believed to be the process underlying the formation of spinal lipomas (F). Complete nondisjunction of cutaneous ectoderm from neural ectoderm results in the formation of myelomeningoceles (Fig. 9-4). Focal nondisjunction results in a persistent epithelium-lined connection between the central nervous system and the skin (G). This persistent connection has been labeled a dorsal dermal sinus. |
rests in the normal filum terminale and distal conus medullaris of the adult are believed to result from the disorder of this process. Several neuronal markers expressed in vertebrate embryos are thought to modulate the differentiation of structures derived from the caudal cell mass and be involved in the maturation of the caudal spinal cord. These include N-CAM, synaptophysin, 3A10, and NeuN (21). The final stage in the formation of the distal spinal cord begins at about 38 days of gestation, at which time the cell mass and central lumen of the caudal neural tube decrease in size as a result of programmed cell death (apoptosis) of the portion derived from primary neurulation (20) and necrosis of the portion derived from secondary neurulation (22). This process has been named retrogressive differentiation (Fig. 9-3). The caudal segment (formed by canalization and retrogressive differentiation) eventually becomes the most caudal portion of the conus medullaris, the filum terminale, and a focal dilation of the central canal (within the conus medullaris) known as the terminal ventricle (ventriculus terminalis) (23,24,25,26,27,28).
 Figure 9-3 Canalization and retrogressive differentiation. After formation of the neural tube, a caudal cell mass forms in the tail fold as a result of fusion of neural epithelium at the caudal end of the embryo with the notochord (A). By the age of 30 days, multiple cysts and clumps of cells appear in the caudal cell mass (B). These cysts coalesce to form a tubular structure that unites with the neural tube above (C). At about 38 days, the cell mass and central lumen of the caudal neural tube decrease in size through cell necrosis in a process known as retrogressive differentiation. The segment formed by this process eventually forms the distal-most conus medullaris, the filum terminale, and the terminal ventricle (D). |
arch (31). Notochordal remnants persist between the newly formed vertebral bodies and become incorporated into the intervertebral discs as the nuclei pulposi. Portions of the thoracic sclerotomes later migrate ventrolaterally to form ribs. The anterior and posterior longitudinal ligaments form during chondrification, from mesodermal cells.
 Figure 9-4 Open spinal dysraphism (myelocele and myelomeningocele). A. Myelocele. The neural placode is a flat plaque of neural tissue that is exposed to the air. The dura is deficient posteriorly; the pia and arachnoid line the ventral surface of the placode and dura, forming an arachnoid sac that is continuous with the subarachnoid space superiorly and inferiorly. Both the dorsal and ventral roots arise from the ventral surface of the placode. B. Myelomeningocele. This is identical to the myelocele with the exception that there is an expansion of the ventral subarachnoid space, which posteriorly displaces the placode. |
 Clinical Manifestations of Spinal Anomalies
Clinical Manifestations of Spinal Anomalies Terminology
Terminologythe simple dysraphic states (fatty and/or fibrous thickening of the filum terminale, intraspinal lipoma, spina bifida, and persistent terminal ventricle) and the complex dysraphic states (split-cord malformation, dorsal dermal sinus, caudal regression syndrome, segmental spinal dysgenesis, neurenteric cyst, and dorsal enteric fistula).
Table 9-1 Clinical-Radiological Classification System for Spinal Dysraphism | |||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||
 Imaging Techniques
Imaging Techniques Abnormalities of Neurulation (Disorders in Which the Spinal Cord Does Not Completely Fuse Posteriorly)
Abnormalities of Neurulation (Disorders in Which the Spinal Cord Does Not Completely Fuse Posteriorly)common, in regions where these modalities are widely available. The ongoing prospective multi-institutional follow-up study will determine the long-term benefits of prenatal repair of this spinal anomaly on neurological and functional outcome.
 Figure 9-5 Prenatal imaging of an open spinal dysraphism (myelomeningocele). A. Sagittal sonogram using a 4-MHz transducer shows a focal loss of the normal echogenicity of the posterior elements of the vertebrae, indicating a bony spina bifida. The meningocele is seen as a hypoechogenic region (arrows) extending dorsally at the level of the spina bifida. B. Higher-resolution image using a 8-MHz transducer shows curvilinear echoes (arrows) representing neural tissue running through the meningocele. C. Sagittal image through the spine shows the lumbosacral spina bifida with the dorsal myelomeningocele (small white arrows). The Chiari II malformation (larger white arrows) is seen at the craniocervical junction. Note the massive hydrocephalus. D. Axial image shows the dorsal bony spina bifida. At this level, the placode (black arrows) is still within the spinal canal. E and F. Extensive cerebellar herniation due to myelomeningocele in a different patient. Image (E) shows an open spinal defect/spinal bifida at the S1 level; no meninges were apparent. Image (F) shows a very small posterior fossa; the black arrow shows a nearly vertical tentorium cerebelli, and the white arrow shows cerebellar tissue extending down to the C5 level. G. Sagittal midline MRI of the same patient as in (E and F) at age 4 months, showing the effects of a prolonged severe CSF pressure gradient. The cerebellum is small and irregular in shape; normal folial patterns cannot be seen. The brain stem is elongated and very narrow, with the pontomedullary junction at the level of C1. Cerebellar tissue extends to the bottom of C4 (white arrow). |
 Figure 9-5 (Continued) |
of the spinal column at the affected level, by the posterior fossa contents extending caudally through the foramen magnum and, in many cases, by ventriculomegaly. In general, when the OSD is uncovered and opens to the amniotic fluid, hindbrain herniation is more severe because the rostrocaudal pressure gradient is increased (Fig. 9-5E-G). Early (prenatal) repair is very important in these cases to minimize these consequences.
 Figure 9-6 Myelomeningocele in a neonate. A and B. Sagittal T1-weighted (A) and T2-weighted images (B) show the spinal cord coursing caudally within the spinal canal to the sacral level. The cord is expanded by syringomyelia (s), with some air (small white arrows) producing some susceptibility artifact within the syrinx. The neural placode (p) extends dorsally through the bony spina bifida. The asterisks (asterisk) mark a wet cloth covering the placode. C. Axial T1-weighted image shows the spinal cord (arrowheads) traversing the subarachnoid space from the spinal canal to the skin surface where it will form a placode. D. Axial T1-weighted image at a lower level shows the placode (p) at its dorsal extent, covered by the wet cloth (asterisk). |
malformation. The spinal cord may be split above (31%), below, (25%) or at the same level (22%) as the OSD (94). In addition to the patients with frank split-cord malformation, 5% of patients with OSD have a duplication of the central canals of the spinal cord cephalic to and at the level of the placode, indicating a mild form of splitting that is insufficient to affect the gross contour of the cord (94).
 Figure 9-7 Hemimyelocele. A. Axial T2-weighted image shows the lower thoracic level split-cord malformation and osseous spur (white arrow) dividing the two canals containing asymmetrical hemicords (black arrows). B. Axial T2-weighted image at a slightly lower level reveals postsurgical changes of a repaired myelomeningocele (now appearing as a flattened placode, white arrow) involving the right-sided hemicord in the thoracolumbar region. The repaired hemicord has retethered at the surgical site (white arrow). The central canal of the left hemicord is dilated (black arrow). C. Coronal T2-weighted imaging through the level of the myelomeningocele repair in this markedly scoliotic patient shows the hypointense osseous spur (white arrow) separating the smaller, slightly hyperintense right hemicord (black arrow), which terminates tethered to the dura surrounded by epidural fat. The larger left hemicord (black arrowheads) continues more caudally. |
 Figure 9-8 Myelomeningocele with split-cord malformation in a 28-week fetus. A. Sagittal RARE imaging shows the lumbosacral posterior osseous defect and dorsal herniation of the placode (black arrow) into and across the myelomeningocele sac. Note also the Chiari II malformation of the brain, including hydrocephalus, reduced subarachnoid spaces, and hindbrain herniation. The lower extremity is gracile with fixed hip hyperflexion, knee extension, and clubfoot deformity (white arrow). B and C. Axial and coronal RARE images demonstrate the dual spinal cords (white arrows in B and C) within the dysraphic lumbar spinal canal, just proximal to the level at which they traverse the posterior osseous defect to enter the myelomeningocele sac (black arrows in B). |
 Figure 9-9 Epidermoid tumor in a patient who underwent fetal myelomeningocele repair. A-D. The epidermoid (arrows) can be difficult to detect by routine sagittal and axial T1 and T2 MR sequences. E. Sagittal FLAIR with fat suppression shows the epidermoid clearly (arrow). Diffusion imaging can also be valuable (see Chapter 7). |
brain stem is pulled/stretched inferiorly and narrowed in the anteroposterior diameter, often lying at the level of the foramen magnum or in the cervical spinal canal. The cervical spinal cord is displaced inferiorly and the upper cervical nerve roots have to ascend toward their respective neural foramina. The medulla is also displaced inferiorly. In 70% of patients, it folds caudally at the cervicomedullary junction, dorsal to the cervical spinal cord (which is tethered by the dentate ligaments and therefore limited in its vertical descent), forming a characteristic cervicomedullary kink (Figs. 5-178 and 5-179). The cerebellar vermis often herniates inferiorly, forming a tongue of tissue posterior to the medulla that usually extends down to the C2 or C4 level. Rarely, it extends down into the upper thoracic segments of the canal. The cerebellum wraps around the brain stem (Fig. 5-179). The fourth ventricle is vertical in orientation (Figs. 5-178 and 5-179), extending inferiorly between the medulla and cerebellar vermis; occasionally, it extends down below the medulla, posterior to the cervical spinal cord, in a cyst-like fashion. The quadrigeminal plate is stretched posteriorly and inferiorly (Figs. 5-178 and 5-179). The small posterior
fossa and the downward herniation of its contents result in a low-lying, abnormally vertical tentorium cerebelli (115,116,117,118). Many of these changes improve or completely resolve after fetal repair of the OSD, as discussed in Chapter 5.
 Figure 9-10 Patient who had myelomeningocele repair at birth, now with worsening neurologic exam secondary to arachnoid scarring/loculations. A. Sagittal T2-weighted image shows the conus medullaris (white arrow) extending caudally to the L5 level. Based on this image, it cannot be determined whether the neurologic symptoms are the result of retethering or another process. This patient emphasizes the need to image the entire spinal cord. The levels of spina bifida (white arrowheads) are those without spinous processes. B. Sagittal T2-weighted image of the cervical and upper thoracic spine shows a thin spinal cord (black arrows) that is dorsally displaced due to scarring and arachnoid loculations. C. Axial T1-weighted image at the midthoracic level shows the spinal cord (white arrows) to have a crescentic shape, being displaced by the scarring and loculations. |
 Figure 9-11 Schematic of dorsal dermal sinus. A tuft of hair, nevus, or hemangioma frequently marks the ostium of the sinus. The dura is often tented when the sinus penetrates the dura. The sinus may terminate in a CNS structure, the dura, or external to the dura. Inclusion cysts develop along the course of the sinus in 50% of affected patients. (Reprinted with permission from Barkovich AJ, Edwards MSB, Cogen PH. MR evaluation of spinal dermal sinus tracts in children. AJNR Am J Neuroradiol 1991;12:123-129.) |
 Figure 9-12 Dorsal dermal sinus with dermoid (inclusion cyst) adjacent to conus medullaris. A. Sagittal T1-weighted image shows the dermal sinus (black arrowheads) coursing through subcutaneous fat and into the subarachnoid space. Part of the subarachnoid portion of the tract is bright (white arrowheads) because it contains fat. The hyperintense circle (white arrow) at the skin surface is a vitamin E capsule marking the opening of the sinus. B. Sagittal T2-weighted image shows the sinus tract (large black arrowheads) more clearly as it courses through the subarachnoid space. Incidentally noted is a thickened filum terminale (small black arrows), likely related to the low level (bottom of L3, one full vertebral body level too low) of the conus medullaris. C and D. Axial T1-weighted images show smudgy soft tissue intensity (white arrows) dorsolateral to the conus medullaris. At surgery, this was found to represent a dermoid. |
diffusion-weighted images can be very helpful in this setting (135,136); they will guide the surgeon to remove as much tumor as possible.
 Figure 9-13 Lumbar dorsal dermal sinus with intramedullary dermoid (inclusion cyst). A and B. Sagittal T1-weighted and T2-weighted images show an intramedullary mass (asterisk) that has similar signal intensity to CSF. Note that the sinus tract is not within the portion of the spine visualized on these images. C. Sagittal T2-weighted image of the lumbar spine shows a hypointense curvilinear structure (black arrowheads) coursing down the dorsal aspect of the subarachnoid space and then terminating (black arrow) at the mid L5 level. As seen in (D), this is the level where the tract exits through the dermal sinus. D. Sagittal T1-weighted image shows the dermal sinus coursing (white arrowheads) below the L5 spinous process and (black arrowheads) through the subcutaneous fat. The ostium is marked by the small white arrow. |
an entity distinct from the myelocystocele. The latter by definition should be an encysted spinal cord. Other authors differentiate cervical myelocystoceles from cervical myeloceles, but consider the latter to be meningoceles (67). However, meningoceles by definition do not contain neural tissue and the cervical myeloceles clearly have a stalk of neural tissue extending through the spinal bifida, as shown by Rossi et al. (141). Cervical (nonterminal) myelocystoceles should also be differentiated from terminal myelocystoceles, which are anomalies of the caudal cell mass and are located at the lumbosacral level. Terminal myelocystoceles are discussed later in this chapter.
 Figure 9-14 Dorsal dermal sinus with infected dermoid inclusion cyst and epidural abscess. A and B. Sagittal T1-weighted images show the importance of proper windowing. In (A), filmed with wide windows, the subcutaneous portion of the sinus (black arrows) is easily seen. In (B), windowed to see the intraspinal portions of the sinus and dermoid, the subcutaneous portion of the tract cannot be identified. In (B), the lower aspect of the thecal sac (arrows) is very heterogeneous. However, it cannot be determined from this study whether the heterogeneous signal is the result of clumping of nerve roots from infection or from the inclusion cyst. At surgery, this patient was found to have a large, infected epidermoid at the L5-S1 levels and an epidural abscess at the L3-L4 levels. C and D. Sagittal T2-weighted (C) and fat-suppressed postcontrast T1-weighted (D) images allow better distinction between the epidural (e) and intradural (i) components of the infection. E. Axial T1-weighted image at the L5 level. The contents of the thecal sac (white arrows) are of heterogeneous high signal intensity. This was found to be an infected dermoid tumor at surgery. The black arrowhead points to the sinus tract coursing through subcutaneous fat. |
motor deficit is usually present, often requiring orthopedic intervention (143). The head size is often enlarged (67).
 Figure 9-15 Contrast enhancement of thoracic dermal sinus. A and B. Sagittal T2-weighted images show the hypointense dermal sinus (white arrows) coursing through the subcutaneous fat and the distortion of the dorsal spinal cord (black arrows) where the sinus tract inserts. C. Sagittal postcontrast T1-weighted image with fat suppression shows the enhancement of the deeper portion of the oblique sinus (white arrow). D and E. Axial T2-weighted images show the lack of complete closure of the dorsal thoracic spinal cord just above the level of the tract (black arrow in D) and the hypointense tract through the subcutaneous fat (black arrows in E). |
much greater near the interface of the cord and lipoma and considerably less near the skin surface (130,146). Calcification and ossification are sometimes seen (130,147), as are muscle fibers, nerves, glial tissue, arachnoid, ependyma, and many other types of tissue (148).
 Figure 9-16 Lumbosacral inclusion cyst following fetal myelomeningocele repair. A. Sagittal T2-weighted FLAIR imaging with fat suppression demonstrates the heterogeneously hyperintense bilobed mass expanding the distal spinal cord (upper two white arrows) and filling the distal thecal sac (lower two white arrows). Note the dorsal osseous defect and the overlying residual soft tissue changes from myelomeningocele and in utero surgical repair. The spinal cord has become retethered by the mass. B. Sagittal thin section steady-state image better reveals the dilated central canal of the lower thoracic spinal cord (arrow), the internal heterogeneity of the inclusion cyst, and the long segment of attachment to the posterior wall of the thecal sac. C and D. Sagittal diffusion-weighted image (C) and apparent diffusion coefficient map (D) show the varying, heterogeneous diffusivity within the proximal and distal portions of the lesion. |
 Figure 9-17 Schematic of myelocystocele. This is an occult, skin-covered, spinal dysraphism in which the spinal cord (which has a syringohydromyelia) and the arachnoid are herniated through a posterior spina bifida. The cyst is in continuity with the central canal of the spinal cord. Myelocystoceles can occur at any level. Localized expansion of the subarachnoid space is not a necessary component and is uncommon in myelocystoceles that occur at locations other than the cord terminus. |
the fat. The faulty disjunction between neural ectoderm and cutaneous ectoderm that results in the formation of spinal lipomas may also explain the frequent association of lipomas with dorsal dermal sinuses, which result from a focal area of faulty disjunction.
 Figure 9-18 Cervical myelocystoceles. A. Sagittal RARE image through the cervical spine in a 22-week fetus shows ventral compression of the cervical spinal cord by the ependyma-lined cyst (asterisk), which extends posteriorly through a spina bifida into the subcutaneous tissues to form a skin-covered dorsal mass (white arrow) covering the neck and occiput. The spinal cord separation due to the cystic dilation of the central canal can be seen traversing the spinal canal (black arrow). B. Axial RARE image shows ventral displacement and compression of the cervical spinal cord (white arrows) by the large myelocystocele (asterisk). C. Sagittal T2-weighted image through the cervical spine of a different patient shows similar compression of the ventral spinal cord (black arrows) by the very large cyst (asterisk), which extends dorsally through the bony spina bifida into the posterior subcutaneous soft tissues. The upper cervical spinal cord is expanded and shows T2 hyperintensity (white arrow), indicating interstitial edema and, likely, a presyrinx state. D. Axial thin section steady-state imaging better depicts the distinction between the intramedullary cyst (asterisk) and the surrounding meningocele components. The malpositioned ventral and dorsal nerve roots can be seen arising from the ventral aspect of the spinal cord bilaterally (white arrows). |
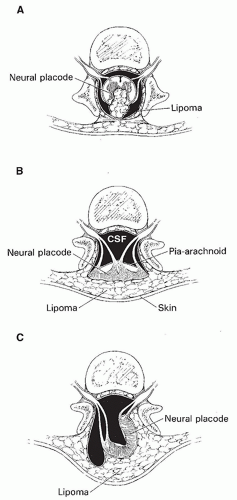 Figure 9-19 Schematic illustrating spinal lipomas and lipomyelomeningoceles. A. Subpial-juxtamedullary lipoma. The spinal cord is open in the midline dorsally with the lipoma situated between the nonapposed lips of the placode. B. Lipomyelocele. This lesion is very similar to a myelocele with two additional characteristics. The lipoma lies dorsal to and is attached to the surface of the placode. This lipoma is continuous with subcutaneous fat. Equally important is the fact that an intact skin layer overlies the lesion, making this an occult spinal dysraphism. C. Lipomyelomeningocele with rotation of the neural placode. When the lipoma is asymmetric, it extends into the spinal canal and causes the ventral meningocele to herniate posteriorly and the dorsal surface of the placode to rotate to the side of the lipoma. This rotation brings the contralateral dorsal root (in this case the right dorsal root) into the midline posteriorly, putting it at increased risk for surgical trauma. Moreover, the left roots are markedly shortened by this rotation, limiting the mobility of the cord and impeding the neurosurgeon from completely untethering it. |
 Figure 9-20 Intradural lipoma CT. A. Axial CT image shows a low-attenuation mass (arrows) filling much of the spinal canal and the interface with the displaced and compressed spinal cord on the left side of the canal. B. Curved sagittal CT reformation of the lumbosacral spine demonstrates the low-attenuation lipoma (black arrows) and multiple osseous anomalies of segmentation (often incorrectly called “fusion”) in the vertebral bodies and posterior elements (white arrows). |
 Figure 9-21 Intradural lipoma MRI. Sagittal (A) and axial (B) T1-weighted images show the hyperintense lipoma (arrows) dorsal to the compressed spinal cord. The spinal canal is expanded by the mass. |
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


