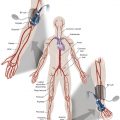
Vascular anomalies are a complex group of developmental abnormalities due to inborn errors of vasculogenesis or angiogenesis that present significant challenges in diagnosis and management. Because of the rarity and diverse presentation of these anomalies, patients are often seen by multiple specialists before correct diagnosis can be reached and treatment instigated. Accurate and timely diagnosis is crucial, and a multidisciplinary team approach is essential for the appropriate evaluation and management.
The cornerstone to understanding the correct diagnostic and treatment algorithm is correct classification and nomenclature. The first classification of vascular anomalies was based on biologic and histopathologic differences described by Mulliken and Glowacki in 1982. The classification broadly separated vascular anomalies into two groups: hemangiomas, where there is abnormal endothelial cell proliferation, and vascular malformations, in which a developmental error has occurred. In 1992 Mulliken and Young founded the International Society for the Study of Vascular Anomalies (ISSVA) and the classification was adopted by the society. At the time there was also the Hamburg classification, a consensus classification of congenital vascular defects published from the seventh meeting of the International Workshop on Vascular Malformations and sixth forum of angiologists in Hamburg in 1988. This classification was based on the anatomic vessel involved and whether the abnormalities were truncal or extratruncular.
More recently it has been recognized that the classification of vascular anomalies is a wide spectrum and advances in genetics, clinical behavior, and histologic entities has significantly increased our understanding and led to a requirement for modification of the original ISSVA classification. This is mainly due to the deeper level of understanding of distinct entities and associated syndromes and the realization of a number of unclassified vascular anomalies under the previous classification. Consequently, a working group was formed by ISSVA and in 2014 at their scientific meeting a revised ISSVA classification of vascular anomalies was approved. This combined some of the aspects of the 1988 Hamburg vascular anomalies classification. ( Table 16.1 ).
| Vascular Anomaly | ||||
|---|---|---|---|---|
| Vascular Malformations | ||||
| Vascular Tumors | Simple | Combined | Of Major Named Vessel | Associated with Other Anomalies |
| Benign | Capillary (CM) | Capillary-venous (CVM) | Affect | Klippel-Trenaunay syndrome (CM + VM ± LM + limb overgrowth) |
| Venous (LFVM) | Capillary-lymphatic (CLM) | Lymphatics | ||
| Locally aggressive | Lymphatic (LFLM) | Capillary-arteriovenous (CAVM) | Veins | Parkes Weber syndrome (CM + AVF + limb overgrowth) |
| Arteriovenous (AVM) | Lymphatic-venous (LVM) | Arteries | ||
| Malignant | Arteriovenous fistula (AVF) | Capillary-lymphatic-venous (CLVM) | Servelle-Martorell syndrome (limb VM + bone undergrowth) | |
| Capillary-lymphatic-arteriovenous (CLAVM) | Anomalies | |||
| Capillary-venous-arteriovenous (CVAVM) | Origin | Limb CM + congenital nonprogressive limb hypertrophy | ||
| Capillary-lymphatic-venous-arteriovenous (CLVAVM) | Course | |||
| Number | Maffucci syndrome (VM ± spindle cell hemangioma + enchondroma) | |||
| Length | ||||
| Diameter | Macrocephaly | |||
| Valves | Microcephaly | |||
| Communication (AVF) | CLOVES syndrome (LM + VM + CM ± AVM + lipomatous overgrowth) | |||
| Persistence (embryonal) | ||||
| Proteus syndrome (CM = VM ± LM + asymmetrical somatic overgrowth) | ||||
| Bannayan-Riley-Ruvalcaba syndrome (AVM + VM + macrocephaly + lipomatous overgrowth) | ||||
Vascular anomalies most commonly present in childhood in one of three ways: a cutaneous lesion, a soft tissue mass, or the result of localized systemic influences of the primary lesion and/or a recognized syndrome. These may be present in isolation or be combined; e.g., a soft tissue mass with a characteristic overlying cutaneous component. Although most vascular anomalies present in childhood, some vascular malformations can present at any age with a preference often for puberty and pregnancy.
Imaging is frequently not required because diagnosis of most vascular anomalies is clinical, especially if there are some characteristic stigmata. Imaging in these circumstances tends to be only for impending critical complications such as periorbital or perispinal anomalies or when the deeper extent cannot be assessed. This is paramount to be able to discuss the best treatment strategy and the likelihood and nature of complications to therapy.
The most useful imaging modalities are magnetic resonance imaging (MRI) and ultrasound (US), including duplex US. US is ideal for outpatient consultations because it is portable, lacks ionizing radiation, is dynamic, and has the ability to detect fluid and flow characteristics. It is also the mainstay of access to allow therapies to be given. MRI offers the best assessment method because it does not utilize ionizing radiation and it has superior tissue contrast resolution and sensitivity to fluid for the evaluation of the extent and tissues involved. For the most part MRI sequences center on identifying static fluid and therefore heavily T2-weighted sequences with and without fat saturation are required along with T1-weighted sequences to identify anatomy. If enhancement characteristics are required to differentiate subtypes of malformations or there is some concern regarding the diagnosis, then postcontrast sequences can be obtained. Postcontrast sequences can be either static or dynamic to allow the understanding of flow. Contrast-enhanced magnetic resonance angiography (MRA) can also be performed if both the arterial and venous anatomy is required. In adults, computed tomographic angiography (CTA) can be used specifically for the anatomy of arteriovenous malformations due to its superior spatial resolution when compared with MRA.
In this chapter, the new 2014 ISSVA classification system is reviewed and described with the typical clinical presentation and imaging findings of each group/subgroup of vascular anomalies. The underlying genetic causes of vascular anomalies are also included in the subsections because this knowledge increasingly plays a pivotal role in target therapies in the management of these conditions.
Vascular anomalies are divided into two main groups: vascular tumors and vascular malformations. Vascular tumors are characterized by clonal cellular proliferation and initial growth in excess of age, whereas vascular malformations are structural abnormalities characterized by errors in vessel morphogenesis present at birth that grow commensurate with age. However, they can grow rapidly at times of hormonal changes (e.g., puberty and pregnancy), and can be complicated by infection, bleeding, and thrombophlebitis. The morbidity and treatment for vascular tumors differ dramatically from vascular malformations, and also differ dramatically for each type of vascular tumor and each type of vascular malformation, which is the overriding impetus for clear, standardized classification in the first place.
Vascular Tumors
Vascular tumors are subdivided into three groups: benign, locally aggressive, and malignant ( Table 16.2 ) and include infantile hemangiomas (IHs); congenital hemangiomas, including noninvoluting hemangiomas (NICHs), rapidly involuting hemangiomas (RICHs) and partially involuting hemangiomas (PICHs); and kaposiform hemangioendotheliomas (KHEs). Age at presentation (prenatal, at birth, early childhood, adult), presence or absence of overlying telangiectatic vessels, a lighter peripheral ring, presence of fast flow, and temporal evolution of the mass (involution vs. no involution) are important clinical criteria to approach the diagnosis of vascular tumors.
| Benign | Locally Aggressive | Malignant |
|---|---|---|
| Infantile hemangioma | Kaposiform hemangioendothelioma | Angiosarcoma |
| Congenital hemangioma | Retiform hemangioendothelioma | Epithelioid hemangioendothelioma |
| Rapidly involuting (RICH) | Papillary intralymphatic angioendothelioma (Dabska tumor) | Others |
| Noninvoluting (NICH) |
Benign Vascular Tumors
Infantile Hemangiomas
Infantile hemangiomas (IHs) make up approximately 90% of all vascular tumors. They are the most common vascular tumors of infancy, with a higher incidence in white infants. The highest incidence is noted in preterm infants weighing less than 1000 g. Head and neck regions are involved most frequently (60% of cases), followed by the trunk (25% of cases) and extremities (15% of cases).
IHs have a characteristic life cycle where they are typically not present at birth but appear commonly within the first 6 weeks of life as a soft, noncompressible mass that grows rapidly in the first few months (proliferative phase) and gradually involutes over years (involutional phase) and remains as a variable amount of smaller fibrofatty tissue for the remainder of life. IHs typically manifest as a single superficial cherry-red “strawberry mark”; deeper IHs are firm, rubbery subcutaneous masses, sometimes with a bluish skin hue. Most IHs double in size in the first 2 months of life, and approximately 80% reach their maximum size by 6 months of age ( Fig. 16.1 ).
Spontaneous regression in early childhood (approximately 7 years of age) is typical, but up to 40% of IHs may have residual skin changes, especially if they involve the head and neck. When a single lesion has a typical clinical history in a territory with no concern, no imaging or intervention is required. However, there are several situations in which further evaluation and treatment may be required. An atypical history requires US evaluation, which typically demonstrates a heterogenous mass with high vascularity on duplex scanning with low resistive arterial waveforms that become increasingly resistant during involution. If five or more subcutaneous lesions are present, then US of the liver is required. MRI reveals a T2 bright, T1 isointense mass with homogenous avid contrast enhancement. Internal serpiginous flow voids within the IH noted in T2-weighted imaging represents the arterial inflow, an important diagnostic clue. Dynamic contrast-enhanced MRA demonstrates early arterial enhancement in a soft tissue mass with a draining vein. Typically, no perilesional edema is observed, which facilitates differentiation from other soft tissue malignancies. Fibrofatty infiltration can be observed during the involuting phase.
Complications of IH include hypothyroidism, heart failure, abdominal compartment syndrome, visual impairment (periorbital), ulceration, and bleeding.
There is an association with more widespread abnormalities. PHACES syndrome refers to a constellation of Posterior fossa brain malformations, Hemangiomas, Arterial anomalies, Coarctation of the aorta and cardiac defects, Eye abnormalities, and Sternal defects.
Patients with hemangiomas overlying the lumbosacral spine can have associated abnormalities, the most common of which is a tethered spinal cord. MRI should be performed to exclude this abnormality. Genitourinary anomalies are possible, although less common.
Airway hemangiomas should be investigated in patients who have cutaneous cervicofacial hemangiomas distributed in the chin, anterior neck, lower lip, and preauricular areas (a “beard” distribution).
Therapy is reserved for those infants in whom there are complications or impingement on vital structures; oral propranolol has largely replaced oral steroids as the therapy of choice, although topical timolol is frequently used for isolated cutaneous lesions. Surgery may be required in some situations. The immunohistochemical marker glucose transporter protein isoform 1 (GLUT1) has become a major tool in diagnosing infantile hemangioma, with endothelial cells staining strongly. Most other vascular lesions, including congenital hemangiomas and vascular tumors, do not stain positive for GLUT1 ( Fig. 16.2 ).
Congenital Hemangiomas
Congenital hemangiomas (CHs) can occur as soft tissue masses, visceral lesions, and intracranial extraaxial lesions. There are three distinct life cycles: RICH ( Fig. 16.3 ), NICH ( Figs. 16.1 and 16.4 ), and PICH. The proliferative phase is complete by birth, although acute enlargement can be secondary to intralesional hemorrhage. They often present as violaceous gray tumors with prominent overlying veins or telangiectases that extend beyond the periphery of the lesion; many have a lighter or bluish halo on the surrounding skin. In RICHs, 90% of involution has occurred at 3 months, with complete resolution by 14 months. NICHs may occasionally grow commensurate with infant growth. Clinically lesions may be associated with a mild consumptive coagulopathy. Imaging is generally not required in CH, but MRI typically demonstrates rim enhancement, which becomes confluent throughout the lesion in time. Histology demonstrates the endothelial cells lining the CH stain negative to GLUT1. Presently no medical therapy has been shown to confer benefit. Surgery is rarely required; however, RICHs can leave significant textural change, necessitating reconstructive surgery after involution.




Tufted Angiomas
Tufted angiomas (TAs) are also known as progressive capillary hemangiomas or angioblastoma of Nakagawa and are cutaneous lesions. TA is associated with Kasabach-Merritt phenomenon, a profound and sustained coagulopathy in which platelets, fibrinogen, and red blood cells are trapped within the lesion. There is speculation that TAs and KHEs are a spectrum of the same entity, with KHE being more aggressive. Histologically TAs contain distinct “cannonball” clusters of packed capillaries and the endothelial cells stain positive for lymphatic marker PROX1.
Pyogenic Granuloma
Pyogenic granuloma (PG), also known as lobular capillary hemangioma, is typically identified in the face, nose, or neck regions as a focal, well-defined superficial lesion of <1 cm. These lesions are extremely vascular and have a tendency to bleed and ulcerate and are associated with a history of recent trauma or inflammation.
Locally Aggressive Vascular Tumors
Kaposiform Hemangioendothelioma
KHE is a rare, distinct vascular tumor. It may present at birth or within the first few months of life as an ill-defined, purpuric mass, often painful, with an encircling pale ring; however, presentation may be later in childhood. The destructive/infiltrative nature and rapid growth of this vascular tumor facilitates differentiation from IH. Kasabach-Merritt phenomenon, a profound and sustained coagulopathy that can be life threatening, can be seen in up to 70% of patients. KHE has a high mortality rate (30%) related to coagulopathy or complications from local tumor infiltration.
KHE can occur anywhere but has a preponderance for extremities and the retroperitoneum. The lesions are firm and indurated and have a purplish coloration in which regional nodal spread has been described without true metastases ( Figs. 16.1 and 16.5 ). KHE appears as a solid mass with ill-defined borders and variable echogenicity on US imaging. MRI demonstrates an infiltrative pattern, with crossing of multiple soft tissue planes and involvement of overlying skin and subcutaneous fat often associated with surrounding edema. Diffuse early enhancement is typical. These more aggressive imaging features distinguish KHE from IH, as do the atypical clinical features.

Histologically KHE consists of coalescing lobules of spindle cells that stain positive for PROX1 (lymphatic marker), CD34 (a stem cell marker), and CD31 (a vascular endothelial marker).
Malignant Vascular Tumors
Angiosarcoma
Angiosarcomas represent 0.5% of all pediatric sarcomas. They can occur anywhere, including viscera—many hepatic cases have been reported. A proportion present with rupture and hemoperitoneum, and 60% have metastases at presentation. Prognosis is poor with a 25% 5-year survival rate. At MRI lesions often contain hemorrhagic products and fluid-fluid levels. At histology angiosarcomas have atypical endothelial cells that stain positive for CD34 and CD31.
Epithelioid Hemangioendothelioma
Epithelioid hemangioendothelioma is an uncommon malignancy that can occur anywhere but is often seen in the liver. The clinical course is more indolent than angiosarcoma, with 5-year survival rates of 73%. At histology the lesions have atypical epithelioid tumor cells with focal intracytoplasmic vacuoles containing red blood cells that stain positive for CD34 and CD31.
Vascular Malformations
Vascular malformations are a complex heterogenous group of developmental abnormalities of vasculogenesis or angiogenesis that present significant challenges in diagnosis and management. The diverse nature of the clinical presentation and the relative rarity of these lesions means that patients have often seen multiple specialists before the correct diagnosis has been made. The lesions are present at birth, although may not present clinically until adolescence or adulthood. They affect males and females equally and persist throughout life. The abnormalities tend to grow slowly, proportional to the patient, and do not demonstrate spontaneous regression. They can occasionally enlarge significantly related to hormonal changes (typically puberty and pregnancy), trauma (including surgery), infection, and thrombophlebitis. The 2014 ISSVA classification now subdivides vascular malformations into simple, combined, of major named vessel, and associated with other abnormalities (syndromes). This chapter focuses on the understanding and clinical features of the simple vascular malformations.
Simple Vascular Malformations
In broad terms a simple way to understand vascular malformations is to subdivide into flow characteristics of low flow and high flow ( Table 16.3 ). A detailed history and examination differentiate vascular malformations into these two groups and from other sinister pathologies. Clinical features are typically related to the focal mass, and patients predominantly present with pain and swelling, which is commonly intermittent. Aesthetic issues are also a common presentation. Clinical examination can separate into high flow (warm, thrill, and bruit) and low flow, which can be subdivided into capillary, venous, and lymphatic ( Table 16.4 ).
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


