Neuromyelitis optica (NMO) is a severe inflammatory demyelinating disorder typically characterized by attacks of recurrent optic neuritis and transverse myelitis. Advances in magnetic resonance imaging techniques and the discovery of the relatively specific NMO IgG biomarker have led to improved diagnostic accuracy and greater recognition of the broad clinical spectrum of aquaporin 4–related autoimmunity. Brain lesions in NMO typically follow the distribution of aquaporin 4 expression and may be symptomatic. Prompt diagnosis of NMO and NMO spectrum disorders has important therapeutic implications given the high risk of recurrent attacks and consequent severe disability, especially in childhood-onset disease.
Key points
- •
Neuromyelitis optica (NMO) is typically characterized by recurrent attacks of optic neuritis and transverse myelitis.
- •
NMO is most common in individuals of female sex and non-Caucasian ethnicity.
- •
NMO IgG is directed against the water channel aquaporin 4 and its presence in serum is 73% sensitive and 91% specific in distinguishing adult-onset NMO from multiple sclerosis.
- •
Increased T2 signal of the optic nerve and/or optic nerve enhancement characterize MR imaging in acute optic neuritis. Longitudinally extensive lesions (>3 spinal segments) are the hallmark spinal imaging finding of NMO. Brain lesions typically follow the distribution of aquaporin 4 expression and may be symptomatic.
- •
Spinal fluid analysis in NMO typically reveals a moderate lymphocytic or neutrophilic pleocytosis. Oligoclonal bands are detected in <10% of children with NMO.
Introduction
Neuromyelitis optica (NMO) is a severe, inflammatory demyelinating disorder of the central nervous system (CNS) that was first described by Devic in 1894. NMO is characterized by recurrent inflammatory demyelination typically restricted to the optic nerve and spinal cord. Historically there has been considerable debate about the relationship between NMO and multiple sclerosis (MS), but NMO is now generally considered a distinct disease with unique clinical, laboratory, and magnetic resonance (MR) imaging features ( Table 1 ). The recent identification of NMO immunoglobin G (IgG) directed against the aquaporin 4 water channel has aided in the recognition of the broad spectrum of aquaporin 4–related autoimmunity and has improved diagnostic certainty.
| Neuromyelitis Optica | Multiple Sclersosis | |
|---|---|---|
| Female:male ratio | Up to 7:1 | 2:1 |
| Relapsing course | 53%–100% | 96% |
| Secondary progression | Rare | Common |
| CSF characteristics | Marked pleocytosis (often >50 cells/μL) Lymphocytes or neutrophils | Mild pleocytosis (typically <30 cells/μL) Lymphocytes |
| CSF oligoclonal bands | 90% | <10% |
| MR imaging brain | Normal, nonspecific white matter changes, or characteristic diencephalic and brainstem lesions | Perventricular, juxtacortical, callosal, with T1 “black holes” indicating chronicity |
| MR imaging spine | ≥3 segments with central/holocordinvolvement | Short-segment and partial cord involvement |
Demographics and epidemiology
Due to its rarity, the precise incidence of pediatric-onset NMO remains unknown. Although generally considered a disease of adulthood, several pediatric case series have now published. In adult-onset NMO, there is a marked female predominance with female-to-male ratio reported as high as 9:1. This striking female predilection is also present in childhood-onset NMO with a female-to-male ratio in pediatric case series reported to be between 2:1 and 7:1. Gender may play a role in influencing NMO disease course; female gender was predictive of a relapsing (as opposed to monophasic) course in 1 large study of adult-onset NMO patients.
Although NMO has been reported in many countries worldwide, its prevalence seems to be the highest in countries with predominantly non-Caucasian populations. In childhood NMO, there is a reported overrepresentation of non-Caucasian ethnicity when compared with other demyelinating diseases such as MS. In a US study of 88 pediatric NMO IgG-seropositive patients, 42 of 58 patients for whom ethnicity was known (73%) were non-Caucasian.
Immunopathogenesis
The recently discovered NMO IgG biomarker is 73% sensitive and 91% specific in distinguishing adult-onset NMO from MS and has similar sensitivity and specificity in children. NMO IgG is directed toward aquaporin 4, a water channel that is expressed on the astrocyte end feet that are a component of the blood-brain barrier. The CNS areas enriched in aquaporin 4 coincide with areas of clinical disease activity (typically optic nerves, spinal cord, brainstem, and diencephalon).
The exact role that NMO IgG plays in disease development has yet to be fully elucidated. Aquaporin 4 is highly expressed in other organs outside of the CNS, including the gut and the kidney, but these organs are generally not clinically affected in NMO patients. Aquaporin 4 is also expressed within the CNS in areas that are often uninvolved (either clinically or radiologically), including the hippocampus, cerebellar granule cells, cerebral cortex, hippocampus, lateral septal nuclei, and the substantia nigra.
However, the high specificity for NMO IgG for a diagnosis of NMO, the association of higher NMO IgG titers during relapses as opposed to remission, and the correlation between antibody titers and cerebral or spinal lesion size all suggest a direct role in disease pathogenesis. Peripherally injected NMO IgG causes exacerbation of disease activity in experimental autoimmune encephalitis, resulting in typical NMO pathologic abnormalities. NMO IgG may be pathogenic by causing internalization of aquaporin 4, complement deposition, altered excitatory neurotransmission, and increased blood-brain barrier permeability.
Clinical course
NMO is typically characterized by the simultaneous or rapidly sequential occurrence of optic neuritis (ON) and transverse myelitis. ON is typically characterized by subacute onset of visual loss (unilateral or bilateral), pain with eye movements, and red color desaturation. Reduced visual acuity (typically 20/200 or worse in NMO), a relative afferent pupillary defect, and optic disc swelling may be detected on physical examination. Transverse myelitis is typically characterized by subacute onset of motor weakness and sensory changes with or without bowel or bladder dysfunction. L’Hermitte symptom (pain with forward neck flexion) may be present with cervical cord involvement. A spinal sensory level is usually detected on physical examination. Myelitis may extend into the brainstem causing hiccups, nausea or vomiting, and respiratory distress.
The clinical course in NMO may be either monophasic (a single attack of ON and transverse myelitis) or relapsing (multiple episodes of ON, myelitis, or attacks involving the brain). In relapsing disease, attacks may be separated by months or years. In children, a relapsing course has been reported in 53% to 100% of patients, although some of these studies had limited follow-up duration. Predictors of a relapsing course in 1 large adult-onset NMO study included female sex, older age at onset, and evidence of systemic autoimmunity. The presence of NMO IgG in serum also seems to be a biomarker for relapsing disease and NMO IgG titer may correlate with disease severity.
NMO is typically characterized by severe attacks and poor recovery, leading to rapid accrual of disability. In 1 large adult-onset NMO cohort study, 47% (31/66) of patients were functionally blind (visually acuity of 20/200 or poorer) in at least 1 eye and 45% (32/71) of patients experienced permanent monoplegia or paraplegia after a mean follow-up time of 16.9 years in the relapsing patients and 7.7 years in the monophasic patients. Respiratory failure was observed in one-third of relapsing NMO patients in this study. In 1 study of childhood NMO, residual motor impairment occurred in 44% (21/44) of patients and persistent visual deficits occurred in 54% (26/48) of patients after a median follow-up time of 12 months.
The identification of the relatively specific NMO IgG biomarker has led to increasing recognition of the broad clinical spectrum of aquaporin 4–related autoimmunity ( Box 1 ). NMO IgG has been found in the serum of patients with recurrent ON and transverse myelitis, which suggests that there may be a continuum of NMO-related disorders ranging from monophasic ON or transverse myelitis to relapsing NMO.
- •
Neuromyelitis optica
- •
Incomplete/partial forms of neuromyelitis optica
- ○
Single or recurrent longitudinally extensive transverse myelitis
- ○
Recurrent or simultaneous bilateral optic neuritis
- ○
- •
Asian opticospinal multiple sclerosis
- •
Optic neuritis or longitudinally extensive transverse myelitis with systemic autoimmune disease
- •
Optic neuritis or longitudinally extensive transverse myelitis with brain lesions typical for neuromyelitis optica (eg, diencephalon or brainstem)
Symptomatic brain involvement
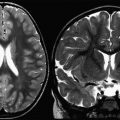
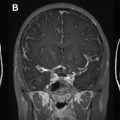
As discussed later in this article, brain MR imaging lesions in NMO typically follow the distribution of aquaporin 4 water channel expression ( Fig. 1 ). Brainstem involvement may present as hiccups, nausea or vomiting, and in severe cases, respiratory failure and death. Diencephalic involvement may manifest as hypersomnolence, narcolepsy, syndrome of inappropriate antidiuretic hormone secretion, or menstrual irregularities. Rarely, coma and death may result from diffuse multifocal cerebral demyelination. Symptomatic brain lesions are thought to occur in less than 15% of adult-onset NMO patients. Symptomatic brain lesions may be more common in childhood-onset disease as demonstrated in 1 study of 42 pediatric NMO patients in which 26 children (45%) had symptoms attributable to brain involvement, including ophthalmoplegia, encephalopathy, seizures, intractable vomiting or hiccups, syndrome of inappropriate antidiuretic hormone secretion, and menstrual dysfunction. In some children, clinical features (polyfocal neurologic deficits and encephalopathy) and MR imaging findings (see later discussion) may resemble acute disseminated encephalomyelitis (ADEM).

Diagnostic criteria
Revised consensus criteria for adult-onset NMO have incorporated the presence of brain lesions and include NMO IgG as a supportive laboratory test. These criteria require the presence of ON, transverse myelitis, and at least 2 of the following supportive criteria: MR imaging evidence of a spinal cord lesion ≥3 segments in length, brain MR images nondiagnostic for MS, or NMO-IgG seropositivity. The presence of 2 of 3 of these supportive criteria confers 99% sensitivity and 90% specificity in distinguishing NMO from MS.
Pediatric consensus criteria for NMO similarly require the presence of ON and transverse myelitis with evidence of either a longitudinally extensive spinal cord lesion or NMO IgG seropositivity ( Box 2 ). Although NMO IgG was present in 78% of children with relapsing NMO in 1 case series, the sensitivity and specificity of the full diagnostic criteria have not yet been formally evaluated in childhood NMO.
- •
Optic neuritis
- •
Transverse myelitis
- •
One of the following:
- ○
Longitudinally extensive spinal lesion (≥3 spinal segments)
- ○
NMO IgG seropositivity
- ○
Systemic autoimmunity
There is considerable debate about the precise relationship between NMO and other systemic autoimmune disorders. There is a high frequency of coexisting autoimmune conditions in NMO patients. Associated autoimmune conditions, including systemic lupus erythematosus, Sjogren syndrome, juvenile rheumatoid arthritis, Graves disease, and autoimmune hepatitis, were observed in 42% (16/38) of children in 1 pediatric NMO case series. Additionally, up to 76% of pediatric NMO patients may demonstrate evidence of serum autoantibodies typically present in other y production even in the absence of a clinical diagnosis of another autoimmune disease. Evidence of systemic autoimmunity may be more commonly found in individuals with relapsing (as opposed to monophasic) disease. Because many patients harbor autoantibodies for other autoimmune conditions apart from NMO without meeting formal diagnostic criteria, the presence of these antibodies may suggest heightened generalized systemic autoantibody production in NMO patients.
Magnetic resonance imaging
MR imaging is an important tool in the diagnostic workup of patients suspected of having NMO and in monitoring disease activity.
MR Imaging Features of Optic Neuropathy in NMO
Patients with ON due to NMO often have bilateral ON and visual acuity typically 20/200 or worse. To date, no MR imaging features have been described that allow differentiation of ON in NMO from ON in MS or other inflammatory causes.
Inflammation and demyelination lead to blood-optic barrier breakdown and nerve tissue edema that can be visualized on MR imaging. In acute ON, fat-suppressed T2-weighted images and short tau-inversion recovery sequences show a hyperintense signal of the affected optic nerve. Fat-suppressed T1-weighted sequences with gadolinium show contrast media enhancement in 94% of patients. Optic nerve enhancement helps to distinguish ON from acute nonarteritic anterior ischemic optic neuropathy, but enhancement alone, however, is not diagnostic for a demyelinating cause. Optic nerve enhancement also occurs in other inflammatory, infectious, or neoplastic diseases. Typical MR imaging features of acute ON are shown in Fig. 2 .

Following acute ON, optic nerve atrophy may occur and may be detected even in patients who have recovered normal visual acuity. MR images in these patients may show persistent abnormalities in short tau-inversion recovery sequences in the absence of contrast media enhancement.
The location and the length of the enhancing optic nerve segment may correlate with the degree of visual loss but is not a predictor of final visual outcome. In a study of 107 patients with acute ON, enhancement >10 mm correlated with more constricted visual fields, and enhancement >17 mm was also associated with significant reduction of visual acuity and color vision at baseline. Visual outcome at 6 months was not correlated with initial enhancement location or length.
MR Imaging Features of Transverse Myelitis in NMO
Longitudinally extensive transverse myelitis (LETM) is the characteristic spinal cord lesion in patients with NMO. Lesions usually extend over more than 3 segments (on average 5.5 segments), but shorter lesions may be observed and do not rule out a diagnosis of NMO. Lesions are usually localized to the cervical and upper thoracic cord in children as well as in adults. In the acute stage, the lesions usually show high signal intensity on T2-weighted images, affecting white and gray matter, and exhibit diffuse uptake of contrast media mainly in the central component of the lesion. The T2 hyperintensive signal surrounds the enhancing part of the lesion, usually interpreted as perifocal edema. Other signal changes including an “eyelike” appearance of the anterior horns mimicking acute ischemia of the anterior spinal artery can occur in patients with NMO ( Fig. 3 ). Contrast media enhancement and diffusion restriction can be seen in both ischemic and inflammatory lesions, although breakdown of the blood-brain barrier in ischemic lesions leading to contrast media enhancement is usually seen after days rather than immediately, as in NMO. In ischemic myelopathy, but not in NMO, edema within the vertebral bodies is expected.